ОРГАНИЧЕСКИЙ СИНТЕЗ
Наука и искусство
М., Издательство "Мир", 2001 г.
|
В. Смит, А. Бочков, Р. Кейпл
ОРГАНИЧЕСКИЙ СИНТЕЗ Наука и искусство М., Издательство "Мир", 2001 г. |
Глава 1 ЦЕЛИ ОРГАНИЧЕСКОГО СИНТЕЗА
Ответить на вопрос, какова роль органического синтеза в науке и практике, совсем не так просто, как это может показаться. При этом, конечно, недостаточно просто сослаться на реальную или хотя бы потенциальную возможность практического использования целевого соединения, даже если в самом широком смысле трактовать понятие “полезность”. Тем не менее, мы сочли необходимым начать данную главу с рассмотрения этого, наиболее очевидного случая, а именно: синтеза органических соединений, практическая полезность которых не вызывает сомнений.
1.1. Цель однозначна и бесспорна
С древних времен человеку были известны чарующие цвета, которые придавали тканям природные красители, добываемые из различных растений и животных. Уже в XIII в. до н.э. финикийцы владели искусством извлечения индигоидных красителей (“тирийский пурпур”) из мантии некоторых средиземноморских моллюсков. Для извлечения 1 г такого красителя требовалось переработать около 10000 моллюсков, что было длительным и трудоемким процессом, и неудивительно, что получаемый продукт ценился в 10—20 раз дороже золота.
В Древнем Риме рецепт производства пурпура относился к категории наиболее тщательно охранявшихся государственных секретов. Согласно указу Нерона, право на ношение одежды пурпурного цвета принадлежало исключительно императору (“королевский пурпур”) [1а]. Подобная романтическая аура продержалась до второй половины XIX в., вплоть до тех времен, когда адепты новорожденной рационалистической науки, органической химии, безжалостно сдернули многовековой покров таинственности и показали, что красящими компонентами природного красителя являются не очень сложные химические вещества, индиго (1) и 6, 6'-диброминдиго (2) (схема 1.1).
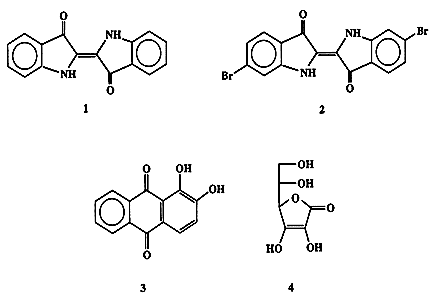
Схема 1.1В 1878 г. Байер [1b] разработал недорогой метод синтеза, пригодный для промышленного производства индиго из доступных исходных веществ. В это же время было охарактеризовано и другое красящее вещество, ализарин (3), выделяемый из корней некоторых видов марены, например, марены красильной (Rubia tinktoria), который с древнейших времен использовался как природный краситель. Первоначально ализарин также был очень дорогим продуктом, однако его цена резко снизилась после того, как Гребе и Либерман [2] предложили в 1868 г. простой способ его получения из углеводорода антрацена, выделяемого из каменноугольной смолы.
Эти поистине триумфальные достижения произвели сильнейшее впечатление не только на химиков, но и на общество в целом. Действительно, трудно было более убедительно продемонстрировать могущество и потенциал органического синтеза, этого едва родившегося, но бурно растущего и дерзкого «младенца».
«Символ веры» молекулярной биологии, «нить жизни» — это ДНК, кодирующая наследственную информацию... Общеизвестная двуспиральная структура этой молекулы была предложена Уотсоном и Криком в 1953 г. Как вспоминал впоследствии проф. Корана, «у меня немедленно появилась честолюбивая мечта синтезировать ее» [3]. Для осуществления этой мечты потребовалось почти два десятилетия напряженнейшего труда большого коллектива, завершившегося блистательным успехом (и Нобелевской премией!) — полным синтезом биологически активного гена — фрагмента ДНК, кодирующего биосинтез тирозиновой транспортной РНК. Это выдающееся достижение не только послужило одним из подтверждений фундаментальных принципов молекулярной генетики, но и явилось мощным толчком к развитию генной инженерии.
Витамин С, аскорбиновая кислота (4), относится к числу важнейших витаминов. Еще в эпоху великих географических открытий человечество столкнулось с роковыми последствиями дефицита в пище этого простого, но тогда еще неизвестного вещества. В те времена несравненно большее число мореплавателей стало жертвами таинственного заболевания, цинги, чем погибло во всех бурях и сражениях. Установление строения аскорбиновой кислоты (1928 г.), а вслед за этим ее лабораторный (Рейхштейн, 1934 г. [4]) и вскоре промышленный синтез из D-глюкозы сделали это вещество дешевым товарным продуктом, тем самым навсегда ликвидировав угрозу цинги (по крайней мере, в нормальных жизненных условиях; реальная, увы, опасность появления этого заболевания в концлагерях любых режимов здесь не рассматривается, ибо причины, обусловливающие такую возможность, лежат далеко за пределами предмета органической химии). Если верить Полингу, то легкодоступный витамин С сможет избавить человека еще от многих других болезней, включая и обычную простуду.
Простагландины (ПГ), такие, например, как ПГЕ1 (5, схема 1.2), впервые выделенные и охарактеризованные в 50-х годах XX столетия, относятся к числу чрезвычайно важных природных соединений. Представители этого до той поры вообще неизвестного класса соединений присутствуют почти во всех тканях млекопитающих и играют ключевую роль регуляторов функционирования таких важнейших систем, как сердечно-сосудистая, системы дыхания, пищеварения и размножения [5а]. Простагландины образуются в организме в микроскопических количествах. Так, в организме взрослого человека синтезируется всего около 1 мг ПГ в день. К сожалению, очень ограничена также возможность их выделения из природных источников. К этому следует добавить высокую лабильность этих соединений, что, конечно, крайне затрудняет их выделение, идентификацию и изучение свойств.
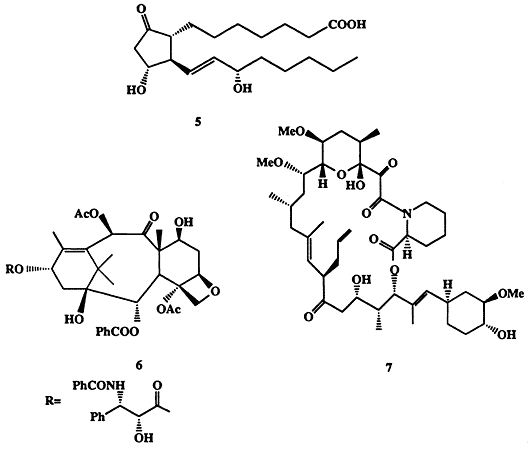
Схема 1.2
Поэтому изначально была очевидна необходимость разработки путей полного синтеза простагландинов, и десятки лабораторий в разных странах начали исследования в этой области. Именно благодаря решению задачи полного синтеза природных простагландинов и большого числа их искусственных аналогов удалось в сравнительно короткий срок добиться действительно впечатляющих успехов как в понимании механизма действия простагландинов, так и в разработке путей их практического использования в медицине и ветеринарии [5b]. Благодаря исключительно высокой активности (в концентрациях порядка нанограмм на 1 мл) как природных ПГ, так и некоторых их синтетических аналогов, синтез этих соединений даже в лабораторных масштабах (от сотен миллиграммов до нескольких килограммов в год) способен удовлетворить потребности целой страны.
“Стоит ли дерево человеческой жизни?” — под таким названием была опубликована статья в американском журнале“Newsweek” (5.09.1991). Под деревом подразумевалась одна из разновидностей тиса, вечнозеленого растения, произрастающего в лесах на западе США и Канады. Одной из специфических особенностей этого растения (собственно и поставивших под вопрос само существование этого вида!) является его способность продуцировать довольно сложную молекулу, таксол (6) (схема 1.2). Это вещество оказалось мощнейшим противораковым средством [6а, b). Оно прошло фазу III клинических испытаний и стало одним из наиболее перспективных лекарственных средств в лечении рака матки и молочной железы, особенно для тех случаев, которые не поддаются лечению другими препаратами.
К счастью, эта дилемма все-таки менее категорична, чем “кошелек или жизнь?” На самом деле существует ряд решений, которые совсем не требуют обязательного уничтожения дерева для того, чтобы спасти человеческую жизнь. Вполне естественно, что сама острота стоящей проблемы послужила мощнейшим стимулом для широкого поиска других природных и возобновляемых источников для выделения таксола и ему подобных веществ, и к настоящему времени стало ясно, что тис не является единственным природным продуцентом соединений этого типа. С не меньшей интенсивностью велись и ведутся исследования по полному синтезу таксола.
Уникальность конструкции углеродного скелета 6 в сочетании с наличием большого числа разнообразных функциональных групп делают синтез таксола исключительным по сложности предприятием. Первые два синтеза соединения 6, выполненные независимо в 1994 г., один — группой Холтона [6с], другой — группой Николау [6d], по справедливости были сразу же отнесены к выдающимся достижениям современной синтетической химии. Оба этих синтеза включают много стадий, и может показаться, что они представляют лишь чисто академический интерес, вне всякой связи с требованиями практики. Однако на самом деле подобные исследования важны еще и потому, что они открывают путь к получению набора разнообразных аналогов целевой молекулы, что необходимо для установления корреляции структура — активность и направленного поиска биологически активных и практически значимых соединений сходного типа, но существенно более простого строения [6b, е].
Головокружительные успехи в области трансплантации органов принадлежат к числу самых ярких демонстраций успехов современной медицины. Безусловно, все это стало возможным в первую очередь благодаря возросшему мастерству хирургов и разработке техники проведения таких операций. Однако не меньшее значение имела также разработка препаратов, способных контролировать иммунный ответ организма пациента с тем, чтобы предотвратить отторжение трансплантируемых чужеродных тканей [7а]. В 1987 г. из культуральной жидкости микроорганизма Strepfomyces tsukubaensis был выделен метаболит FK-506, который оказался одним из наиболее эффективных иммуномодуляторов и для которого было установлено строение макро-циклическоголактона 7 (см. схему 1.2) [7b].
Менее чем через 2 года полный синтез структуры 7, несмотря на устрашающую ее сложность, был завершен группой Шинкаи лаборатории фирмы Мерк Шарп и Дом [7с]. Безусловно, этот синтез не может всерьез рассматриваться как альтернатива в общем-то довольно дешевому процессу микробиологического синтеза. Но именно благодаря синтетическим усилиям в этой области удалось разработать методы получения ряда родственных соединений и изотопно меченных аналогов 7 [7d-f]. Это, в свою очередь, обеспечило возможность проведения исследований, направленных на выяснение особенностей взаимодействия иммуномодуляторов с рецепторами соответствующих клеток, т.е. тех особенностей, без знания которых невозможен рациональный дизайн иммунодепрессантов, более простых по строению, чем макроциклическийлактон 7, но проявляющих требуемый спектр свойств.
Не представляет труда умножить многократно число примеров, свидетельствующих об огромном вкладе достижений органического синтеза в создание современной цивилизации, — вкладе, затрагивающем буквально все аспекты нашей повседневной жизни. Различна техническая сложность этих синтезов и их масштабы — от миллиграммов до миллионов тонн, а также значимость для жизни человечества. Однако что бы не являлось объектом синтеза — будь то синтетические волокна и каучуки, лекарства и красители, высокооктановый бензин или моющие средства, витамины, гормоны или реагенты для различных целей — во всех случаях их целью является получение веществ с практически полезными свойствами и, стало быть, вряд ли могут возникнуть сомнения в целесообразности синтетических исследований в этих областях. Столь ясно выраженная практическая направленность этих работ в сочетании с очевидной полезностью их конечных результатов, пожалуй, в наибольшей степени отвечают ожиданиям той части налогоплательщиков, которые желали бы максимально быстро получать ощутимо полезную отдачу от денег, вложенных в развитие науки.
1.2. Цель однозначна, но не бесспорна
Однако важность того или иного направления в науке чаще всего не может быть оценена столь прямолинейно только по критерию немедленной полезности конкретных научных исследований. На протяжении всей истории органической химии синтетики стремились синтезировать самые различные соединения, выделяемые из живых организмов, часто вне видимой связи с их реальной или хотя бы потенциальной полезностью. Это очень устойчивая тенденция и можно утверждать, что она только усилилась за последние десятилетия. Если раньше иногда приходилось затрачивать десятилетия на осуществление таких синтезов, то сейчас разрыв между открытием нового природного соединения (а такие открытия совершаются в буквальном смысле слова ежедневно) и его полным синтезом сократился до немногих лет, а иногда и месяцев. Законно спросить, зачем делаются подобные синтезы, в чем смысл и значение получения в лаборатории вещества, которое уже было синтезировано в Природе?
Конечно, справедливо утверждение, что во многих случаях сложность и нетривиальность структур природных соединений воспринимаются сами по себе как вызов созидательным способностям Человека и, конечно, для ученого невозможно не принять этот вызов. “Почему Вам столь необходимо взойти на Эверест?” — спросили Мэллори, легендарного английского альпиниста 20-х годов. “Потому, что он есть!” — был ответ. Однако помимо подобного, может быть несколько романтического, стимула, существует также еще вполне конкретная мотивация необходимости синтеза природных соединений. Дело в том, что живая Природа ничего не делает просто так, для “забавы” — ведь биосинтез всегда сопряжен со значительными энергетическими затратами и, если его результатом является получение абсолютно бесполезного соединения, то организм-продуцент не имеет шансов выжить в процессе естественного отбора.
Поэтому смело можно принять как аксиому, что все, что синтезируется организмами, так или иначе связано с обеспечением их жизненно важных функций, и, следовательно, не может не быть важным и для жизни человека. Это может показаться слишком общим или даже банальным утверждением, но на самом деле по мере эволюции наших знаний нам постоянно предъявляются все новые и вполне конкретные доказательства того, насколько оправдано такое отношение к химии природных соединений. Покажем это на некоторых примерах.
Среди множества природных соединений существует обширный класс изопреноидов (или терпеноидов), включающий тысячи структурно различных соединений, которые объединены единством пути биосинтеза из небольшого числа ключевых предшественников. Роль некоторых соединений этого класса, таких, как витамины А и D или стероидные гормоны, уже давно известна: они выполняют важнейшие регуляторные функции в организмах млекопитающих. Также понятна практическая полезность ряда других издавна известных изопреноидов, как, например, камфоры, ментола или каучука. Однако долгое время ничего конкретного не было известно ни о функциях, ни о полезных свойствах еще сотен природных соединений этого класса. В результате к 50-м годам XX в. сложилось мнение, что большинство изопреноидов, например растительного происхождения, образуются в живой клетке как физиологически инертный балласт для связывания отходов метаболизма (вторичные метаболиты).
При этом как-то даже не ставился такой вопрос: а почему все-таки организму потребовалось ценой значительных затрат энергии синтезировать те или иные, иногда очень сложные структуры, если их единственное назначение — обеспечивать функционирование системы удаления “шлаков”? В те времена могло показаться, что лишь профессиональный педантизм и отсутствие воображения заставляют химиков вести нескончаемую работу по поиску и выделению, изучению строения, а также еще и синтезу все новых и новых природных изопреноидов. Типичная “инвентаризация неликвидов, числящихся на балансе природы” — вот мнение, которое авторам доводилось слышать от некоторых ученых-функционеров, от которых, к сожалению, зависело распределение средств на научные исследования.
Однако уже в 1960-х годах эти представления пришлось радикальным образом пересмотреть. В частности было установлено, что не только млекопитающие, но насекомые и даже растения широко используют изопреноиды в качестве гормонов. Так, например, одно из поразительнейших биологических явлений, метаморфоз насекомых — возникновение взрослой особи из личинки через ряд промежуточных стадий, — контролируется тщательно согласованным действием ряда гормонов, вырабатываемых соответствующими железами. Одна из таких парных желез, так называемые прилежащие тела (corpora allata), выделяет ювенильный гормон (ЮГ) (8, схема 1.3), который способствует развитию личиночной стадии насекомого. В определенный момент секреция этого гормона падает и резко возрастает секреция другого гормона, экдизона (9), вырабатываемого проторакальными железами, что прекращает рост личинки и «запускает» последующие стадии метаморфоза, приводящие в конечном счете к взрослой особи. Если в этот момент в организм личинки ввести извне некоторое количество ЮГ, то метаморфоз не наступает, и она продолжает просто расти в размерах, давая в конце концов гигантскую, но нежизнеспособную особь.
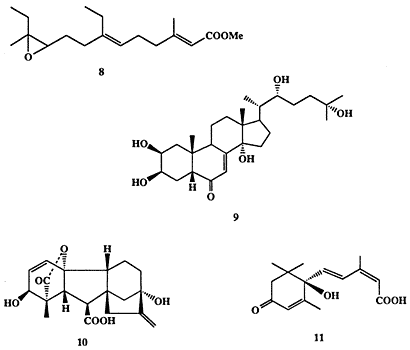
Схема 1.3Оба эти гормона (8 и 9) являются модифицированными изопреноидами [8а] Самый богатый природный источник ЮГ (8) - брюшной сегмент самца бабочки павлиноглазка Hyalphora cecropia - содержит не более 1-2 mг на особь Тем не менее, 8 довольно быстро стал доступным соединением благодаря тем огромным усилиям, которые были затрачены на его полный синтез [8b] Выяснение роли и успешный синтез ЮГ вызвали целую лавину исследований направленных не только на изучение механизма действия этого вещества но и на создание многочисленных его синтетических аналогов, некоторые из которых нашли практическое применение как инсектициды нового поколения.
Некоторые терпеноиды растений также выполняют жизненно важные функции гормонов роста и развития. Так, из метаболитов Glbberella fujikuroi - гриба, вызывающего аномальный рост и гибель рисовых побегов - был выделен дитерпен гибберелловая кислота (10). Вскоре было найдено, что кислота 10 и ее многочисленные аналоги, гиббереллины (известно свыше 40 природных соединений этого типа), широко распространены среди высших растений [9а]. Наиболее характерный физиологический эффект гиббереллинов — ускорение роста стебля за счет как деления, так и растяжения стенок клетки. Но, как и в случае большинства других гормонов, действие гиббереллинов не ограничивается только этим эффектом, и они принимают участие еще во множестве событий развития и роста растений. Синтез гиббереллина 10, выполненный группой Кори [9b], относится к числу выдающихся достижений органической химии. Хотя этот синтез включал более 40 стадий и, конечно, не имеет перспектив практического использования, но вряд ли кто-нибудь может усомниться в его целесообразности, поскольку целью синтеза было получение соединения, являющегося одним из важнейших регуляторов жизнедеятельности организмов всего царства растений.
Другой терпеноид, абсцизин (11) [10], также выделенный из большого числа растений, играет роль антагониста гиббереллина. Действительно, было обнаружено, что в присутствии 11 подавляется рост побегов и инициируется образование “спящих” почек. Таким образом, переход от фазы активного роста в условиях длинного дня к фазе “сна” в условиях короткого дня контролируется балансом содержания в клетках соединений 10 и 11.
Богатейший ассортимент изопреноидов обнаружен среди метаболитов растений и грибов. Среди них встречаются очень сильные токсины, соединения с противоопухолевой и противовоспалительной активностью и антибиотики различного спектра действия. Как правило, очень мало или вообще ничего неизвестно о том, какова роль этих веществ в жизнедеятельности организмов-продуцентов. Однако тот факт, что соединения этой группы проявляют столь широкий спектр биологической активности по отношению к другим биологическим объектам, не может не рассматриваться как указание, по крайней мере косвенное, на их участие в регуляции каких-то биологических функций, существенно важных для организмов-продуцентов. Это утверждение может показаться чисто декларативным, но далее мы приведем примеры того, какие нетривиальные биологические функции могут быть обнаружены, если рассматривать тот или иной организм не как изолированную особь, как это делалось еще сравнительно недавно, а во всем комплексе его взаимоотношений с другими членами биологического сообщества, т. е. как часть экосистемы.
Ни один из живых организмов не может существовать вне связи с другими живыми существами, образующими данный биоценоз. Поэтому для действительного понимания функций природных соединений необходимо рассматривать также возможность их участия в качестве медиаторов, регулирующих тем или иным образом взаимоотношения данной особи как с особями того же вида, так и с совершенно иными организмами данного сообщества. Пока мы находимся еще в самом начале пути к пониманию этих важнейших аспектов химической экологии. Тем не менее уже накопилось множество фактов, однозначно свидетельствующих о наличии и жизненной важности функционирования химического канала связи на всех уровнях организации биологических систем.
Для соединений, выступающих в роли химических сигналов (т.е. переносчиков информации между организмами), предложен термин феромоны или экзогормоны (более общий английский термин — semiochemicals). В роли такого рода сигналов в живой природе могут использоваться соединения практически любых классов, различных по сложности и характеру функционализации, в том числе и изопреноиды, о которых шла речь выше. Ниже будут рассмотрены некоторые типичные примеры, призванные показать разнообразие структур этих веществ и выполняемых ими функций.
По-видимому, именно насекомые за время своей эволюции смогли выработать наиболее изощренную систему химической сигнализации, играющую огромную роль в их жизни. Действительно, с помощью феромонов они способны передавать информацию о присутствии особей того же или иного вида (сигналы узнавания наподобие армейских сигналов опознания “свой — чужой”), о местонахождении самца или самки (половые аттрактанты), о приближении опасности (феромоны тревоги), о наличии источника пищи и маршруте к нему (феромоны агрегации и маркеры следа) и о многом другом.
Эффективность взаимодействия между особями насекомых на больших расстояниях действительно поразительна. Самка бабочки тутового шелкопряда Bombyx mori испускает аттрактант, бомбикол, который способен вызывать ответную реакцию самца на расстоянии до 10 км!
Невероятно трудная работа по выделению этого соединения была выполнена Бутенандтом и сотр. [11а], которым после нескольких лет кропотливой работы удалось выделить всего лишь 3 мг бомбикола путем обработки 31000 желез, каждая из которых была удалена из девственной самки Bombyx mori [[11а]]. Структура этого вещества оказалась на удивление простой — (10E, 12Z)-гексадекадиен-10, 12-ол-1 (12) (схема 1.4), и его синтез потребовал всего лишь нескольких недель [11b].
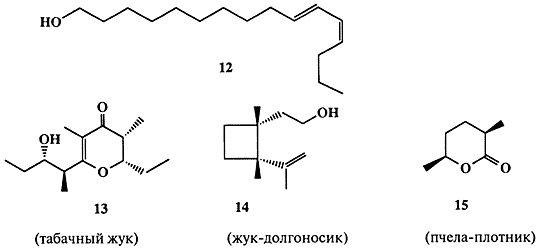
Схема 1.4
Половые аттрактанты многих других бабочек и мух тоже, как оказалось, имеют довольно простую структуру. Это, как правило, ациклические спирты с длинной неразветвленной цепью. Другие насекомые продуцируют в качестве половых феромонов соединения более сложной структуры, некоторые из которых (13-15) показаны на схеме 1.4 [12].
Массированные атаки насекомых могут приводить не только к полной гибели конкретного растения, но и катастрофическому опустошению лесов и полей и уничтожению продовольственных запасов на складах. Эти атаки обычно провоцируются феромонами агрегации, которые испускаются особями, обнаруживающими запасы пищи. Вот какова, например, схема (конечно, очень упрощенная!) такой атаки, используемая жуком-лубоедом Dendroctonus brevicomis — вредителем сосновых лесов. Самка этого жука, найдя подходящее дерево, затем привлекает к себе самцов с помощью аттрактанта, в роли которого выступает бревикомин (16, схема 1.5). Освоивши дерево, «колонизаторы» начинают выделять смесь феромонов, в состав которой входят изопреноиды 17-19, что воспринимается находящимися вблизи особями как приглашение «разделить обильное угощение». В конечном результате популяция вредителей быстро увеличивается в сотни раз и обреченное дерево вскоре погибает [12].
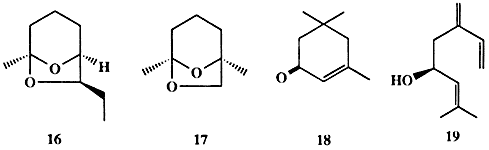
Схема 1.5
Функционирование канала химической информации особенно важно для поддержания жизнедеятельности коллективных насекомых. Почти идеальный порядок социальной жизни пчелиного улья поддерживается до тех пор, пока пчелиная матка сохраняет способность вырабатывать довольно простое соединение, (E)-8-оксодецен-2-овую кислоту (20) («королевское вещество») (схема 1.6) [13a, b].
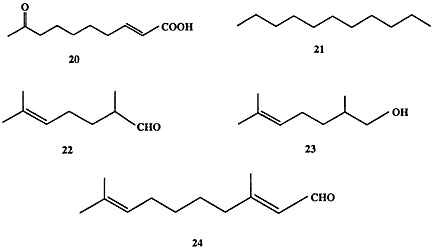
Схема 1.6
Это вещество является химическим сигналом, выполняющим функцию многоцелевого регулятора. Во-первых, он служит половым аттрактантом для самцов, что обеспечивает постоянную возможность воспроизведения обитателей улья, а во-вторых, он, по-видимому, является чрезвычайно желанным «деликатесом» для остальных членов пчелиного семейства, которые не упускают случая им полакомится. Однако в результате такого «угощения» подавляется как репродуктивная функция других самок, так и их инстинкт к строительству ячеек большего размера, необходимых для выкармливания новой матки. Благодаря этому в улье в принципе невозможны ни «государственный переворот», ни даже появление нового «претендента на власть» до тех пор, пока матка остается дееспособной, т. е. сохраняет способность вырабатывать королевское вещество 20.
Строгая иерархия муравьиной колонии жестко определяет место и специализацию каждого из ее членов. Примечательные способности муравьев в организации “коллективных работ”, таких, как сооружение муравейника, походы за пищей или с целью захвата “рабов”, культивация грибов или содержание тлей в качестве “домашних животных”, не могут не вызывать восхищения.
Накоплено множество данных, позволяющих утверждать, что одним из основных инструментов в организации подобного рода согласованных действий членов муравьиного сообществ является коммуникация между отдельными особями с использованием химического канала связи. Химические сигналы являются медиаторами на уровне как “вертикальных”, так и “горизонтальных” связей.
Например, так называемые кастовые феромоны, вырабатываемые маткой муравейника, определяют будущую специализацию молодой особи — “работника” или “воина”. В последнем случае, кроме мощных челюстей, особь наделяется также способностью вырабатывать феромон тревоги. Попадание в среду этого феромона даже в незначительном количестве немедленно вызовет цепную реакцию его выделения другими “солдатами”, происходит многократное усиление сигнала тревоги, и в течение нескольких секунд колония муравьев полностью приводится в состояние боевой готовности. При других условиях феромон тревоги может также провоцировать состояние паники и даже бегства всей колонии.
С помощью других сигналов, феромонов агрегации, “работники” сообщают о необходимости включиться в строительство в определенном месте. Задачей некоторых “солдат” является разведка новых источников пищи и обнаружение целей для “грабежа”. Естественно, что выполнение этой задачи требует использования феромонов, маркеров следа, причем эти сигналы должны быть достаточно специфичны, чтобы сообщения были понятны лишь членам данного семейства. Как правило, довольно простые по структуре вещества и их смеси используются муравьями в качестве химических сигналов. Так, например, в роли феромонов тревоги ряд видов муравьев использует набор веществ типа 20-24 (схема 1.6) в варьируемых соотношениях [14].
Необыкновенно разнообразен набор природных веществ, продуцируемых низшими растениями. Биологические функции большинства из них просто неизвестны. Однако и для низших растений показано, что многие метаболиты выполняют функции медиаторов взаимоотношений между индивидуальными особями.
Интересно, что первые догадки о наличии таких химических сигналов высказывались наблюдательными натуралистами еще в середине XIX в. Так, в 1858 г. было предположено, что в цикле полового размножения морской водоросли Fucus serratus мужские половые клетки (андрогаметы) сближаются с женскими половыми клетками (гиногаметами) благодаря наличию в среде химических сигналов (хемотаксис) [15а]. Прошло более 100 лет, прежде чем удалось доказать справедливость предположения о роли продуцируемых и испускаемых в среду химических веществ как важнейших медиаторов в половом размножении. Оказалось, что для этой водоросли таким медиатором является триеновый углеводород фукозосерратен, C8H12 (25, схема 1.7), который продуцируется женскими половыми клетками и при попадании в водную среду служит половым аттрактантом для андрогамет. Подобный механизм широко используется и другими видами низших растений. Структуры 26-28 [15a, b] наглядно иллюстрируют, насколько разнообразные по строению углеводороды могут продуцироваться разными организмами для выполнения одной и той же функции.
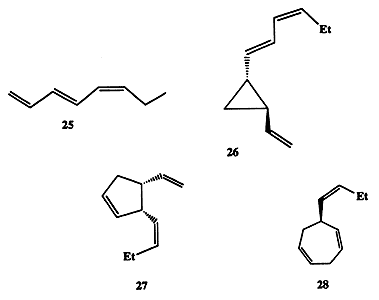
Схема 1.7
Разумеется, все эти углеводороды практически нерастворимы в воде, что делает еще более интересным факт их фантастически высокой эффективности. Так, установлено, что положительный отзыв на сигнал может наблюдаться уже при пороговой концентрации 6, 5 x 10-12 моль/л. Расчет показал, что для привлекающего эффекта достаточно, чтобы на одну андрогамету приходилось от одной до десяти молекул феромона[15а].
Для женских гамет некоторых видов гриба рода алломицетов Allomyces, обитающих в пресных водах, оказалось почему-то предпочтительнее вырабатывать в качестве полового аттрактанта сесквитерпен сиренин (29) [16]. Интересная последовательность событий, каждое из которых контролируется соответствующими экзогормонами, наблюдалась в цикле полового размножения морской водоросли Achlia bisexualis. В этом процессе само формирование мужских и женских органов, предшествующее акту размножения, происходит в результате обмена сигналами, выделяемыми в среду. Интересно, что одним из веществ, используемых как сигнал в этих “переговорах”, является стероид антеридиол (30) [17]. Этот, казалось бы частный, факт на самом деле является примечательным прежде всего потому, что он относится к числу свидетельств, пока еще немногочисленных, указывающих на то, что стероиды могут выполнять не только функции регуляторов жизнедеятельности внутри данного организма (эндогормоны), но также служить регуляторами взаимоотношений на межорганизменном уровне (экзогормоны).
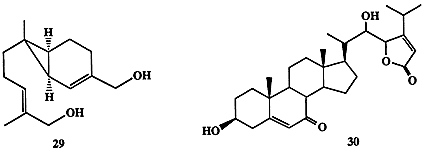
Схема 1.8
Факты, свидетельствующие о сложных, а подчас даже таинственных, взаимоотношениях между различными организмами, образующими биоценоз, известны с давних времен. Во многих случаях было также очевидно, что эти взаимоотношения каким-то образом связаны с химическими веществами, выделяемыми в среду. Однако лишь сравнительно недавно стали накапливаться строгие экспериментальные данные, свидетельствующие о существовании химического канала обмена информацией между особями разных видов. Здесь мы находимся еще в самом начале пути к пониманию всей картины взаимоотношений. Тем не менее ряд приводимых ниже фактов достаточно четко показывают важность функционирования химической сигнальной системы для жизни биологических сообществ.
Семена растения-паразита Striga asiatica (witch-weed, “ведьмин сорняк”) могут находиться в почве в состоянии спячки до 20 лет, но они немедленно начинают прорастать, как только поблизости от них появляются корни растения-хозяина. Этот факт не является просто еще одним примером из огромного списка любопытных биологических феноменов. Дело в том, что растение-хозяин — это рис, сорго, а также ряд других злаков, и поэтому засоренность посевов этих культур таким сорняком может нанести огромный ущерб урожаю, от которого зависит жизнь миллионов людей. Каким же образом семена растения-паразита “узнают” о том, что рядом находятся прорастающие семена злака, и как выбирают правильное направление роста с тем, чтобы в конце концов прикрепиться к корням растения-хозяина?
Изучение этой проблемы показало, что прорастание семян сорняка провоцируется химическими веществами, вырабатываемыми растущими корешками растения-хозяина, и эти вещества служат факторами прорастания для семян растения-паразита.
Одним из таких стимуляторов роста оказался терпеноид стригол (31, схема 1.9) [18а]. Естественно было предположить, что предпосевная обработка полей этим соединением будет вызывать прорастание семян сорняка и тем самым откроет возможность радикального решения проблемы борьбы с ним. Неудивительно поэтому, что многочисленные исследования были направлены на разработку удобного метода синтеза соединения 31 и его более простых аналогов [18b]. Впоследствии было обнаружен еще один активный стимулятор прорастания семян растения-паразита, также вырабатываемый растущими семенами злаков рода Sorghum, а именно замещенный гидрохинон 32 [18с]. Подобно другим гидрохинонам вещество 32 способно легко претерпевать окисление, давая хинон 33, и этот процесс легко протекает в почве. Хинон 33 не является стимулятором прорастания семян сорняка, и поэтому прорастание последних может произойти лишь в том случае, если семя растения-паразита находится на достаточно близком расстоянии к растущему корешку растения-хозяина, поскольку в противном случае стимулятор 32 успеет полностью превратиться в неактивный 33 в ходе диффузии через почву.
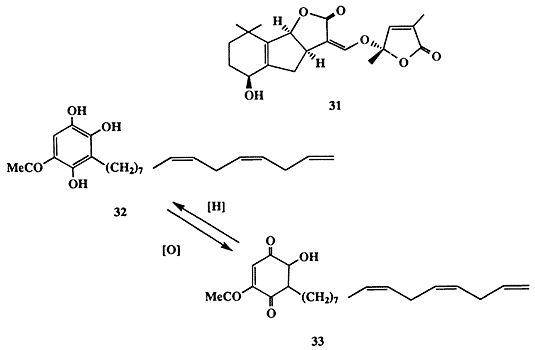
Схема 1.9
С “точки зрения” растения-паразита, это, конечно, очень удобный механизм, поскольку длина его растущих корешков не превышает 3 мм, передвигаться в почве семена не могут, и поэтому им нет никакого смысла подвергаться активации на большем расстоянии. Вопрос о том, как и для чего растение-хозяин обзавелось столь самоубийственной способностью способствовать росту своего паразита, по-видимому, следует отнести к одной из многочисленных загадок эволюции.
Теснейшая взаимосвязь между растениями и насекомыми — хорошо изученный биологический феномен, и накоплено множество фактов, указывающих на огромную роль химических веществ как регуляторов этих взаимоотношений [19]. Примерно полмиллиона видов насекомых кормится на растениях. В свою очередь, процессы репродукции множества растений критически зависят от переноса пыльцы, осуществляемого насекомыми. Поэтому неудивительно, что среди множества природных веществ, продуцируемых растениями, можно найти как аттрактанты для “полезных” насекомых, так и репелленты или даже инсектициды для “вредных” [20]. Фантастическое разнообразие структур соединений, используемых для этих целей (среди них можно найти ациклические и полициклические соединения, в том числе изопреноиды, ароматические и гетероароматические соединения, множество алкалоидов различного строения и т. д.) может служить прекрасной иллюстрацией того, насколько широки возможности Природы в выборе структур органических соединений, выполняющих те или иные функции. Однако надо сказать, что в общем имеется немного достоверных сведений о реальном механизме действия химических медиаторов во взаимоотношениях растений и насекомых.
Один из интереснейших примеров, иллюстрирующих некоторые аспекты химических взаимоотношений между растениями и животными можно найти в работах группы Мейнвальда [21]. Алкалоиды (как и терпеноиды) относятся к числу так называемых вторичных метаболитов, т.е. веществ, не принимающих участия в основных циклах метаболизма. Эти азотсодержащие соединения в значительных количествах продуцируются различными растениями. Многие из этих соединений обладают ярко выраженной активностью по отношению животным (общеизвестна активность, например, морфина или стрихнина), но роль большинства алкалоидов в обеспечении жизнедеятельности организма-продуцента пока совершенно неизвестна.
Сравнительно недавно было установлено, что во многих случаях они выполняют функции защиты растения от поедания насекомыми (антифиданты). Однако эта защита, как правило, оказывается не универсальной, поскольку в ходе эволюции некоторым видам насекомых удалось выработать устойчивость по отношению к этим ядам, а иные “ухитрились” разработать довольно изощренную схему использования потребляемых с пищей алкалоидов в своей жизнедеятельности. Так, например, личинки бабочки Utetheisa ornatrix способны поедать листву растения семейства Crotolaria, поглощая при этом в довольно больших количествах ядовитые пирролизидиновые алкалоиды, в том числе монокротоналин (34, схема 1.10). Как было обнаружено, такая диета дает дополнительные преимущества для выживания вида, поскольку накопление 34 делает несъедобными и личинки, и взрослые особи для таких энтомофагов, как пауки или птицы [21].
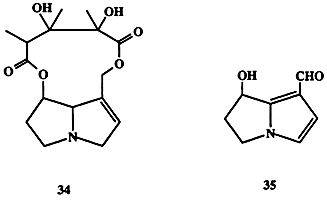
Схема 1.10
Во взрослых организмах U. ornatrix 34 частично превращается в другой алкалоид гидроксиданаидаль (35), который также обладает свойствами антифиданта для энтомофагов, являясь в то же время составной частью секрета, продуцируемого самцами бабочки в качестве аттрактанта. Чем больше содержится этого вещества в секрете, тем выше шансы у самца найти партнера для спаривания. Причина подобного предпочтения довольно понятна: высокое содержание 35 в аттрактанте гарантирует высокое содержание 35 в семени самца, и, следовательно, значительная часть этого вещества будет перенесена в отложенные яички, которые благодаря этому станут несъедобными для таких врагов, как божьи коровки. Эволюционные преимущества подобной “заботы” о потомстве очевидны.
У читателя может возникнуть вполне законный вопрос: зачем в книге, посвященной органическому синтезу, рассматривать примеры, хотя и интересные, но скорее относящиеся к общей биологии? Для нас самый главный вывод из рассмотренных выше примеров (а их число легко многократно умножить) состоит в том, что при решении вопроса о биологической активности или функции того или иного природного соединения нельзя ограничиваться только рассмотрением его возможных функций в организме-продуценте или его свойства как потенциально полезного лекарства. На самом деле адекватное рассмотрение этой проблемы требует системного подхода, учитывающего сложность взаимоотношений внутри биологических систем на всех уровнях организации — от клеток до биоценозов.
Рассмотренные выше примеры относятся к довольно простым случаям, когда химические вещества служат медиаторами простых и хорошо определенных взаимоотношений внутри организма или между немногими особями. На самом деле взаимоотношения между биологическими партнерами образуют сложно переплетенную сеть горизонтальных и вертикальных связей, охватывающих все сообщество. Стабильность интегрированной биологической системы как единого целого критически зависит от взаимодействия отдельных ее частей. Есть все основания предполагать, что химический канал связи в действительности является одной из важнейших составных частей системы контроля, обеспечивающего эффективность этого взаимодействия, хотя до сих пор мы не имеем целостного представления о системе химической коммуникации в сколько-нибудь сложных биологических сообществах.
Очевидно, что знание языка химической коммуникации (словаря сигналов) и его синтаксиса совершенно необходимо для установления разумных и взаимополезных отношений человека с окружающей живой природой. Это позволит нам остановить, наконец, бесконечные и бессмысленные войны с Природой в попытках истребить “вредные для человека” виды и перейти к диалогу на универсально понятном языке химических сигналов и, следовательно, к разумной регуляции наших отношений с другими живыми существами. Это, конечно, не более чем очень далекая перспектива, но сама возможность движения в этом направлении во многом зависит от успехов органического синтеза.
В заключение раздела отметим еще, что название раздела “Цель однозначна, но не бесспорна”, конечно, достаточно приблизительно. Когда речь идет о синтезе природного соединения, то, даже если неизвестно, для каких целей оно синтезируется живым организмом и отсутствуют данные о его биологической активности, сам факт выделения такого соединения из природных источников делает бесспорной необходимость его лабораторного синтеза. Подобная категоричность может, конечно, вызвать множество возражений, но вся история развития химии природных соединений представляет множество свидетельств в пользу справедливости этого тезиса. В подтверждение приведем лишь один пример.
В 1968 г. из культуры гриба Pseudorotium ovalis был выделен сесквитерпен (-)-овалицин (36, схема 1.11). Хотя этот метаболит проявлял ряд свойств антибиотика, он не казался перспективным в качестве лекарственного средства и, как это бывает достаточно часто, не имелось абсолютно никаких данных о его возможной роли в жизнедеятельности организма-продуцента. Тем не менее, необычность и сложность строения этого соединения не могли не вызвать у синтетиков амбициозного желания синтезировать эту структуру.
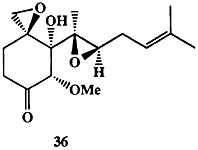
Схема 1.11
Синтез рацемического 36 был выполнен Кори в 1985 г. [22а], и одной из его целей была просто проверка предлагаемых общих принципов методологии органического синтеза. Схема синтеза включала последовательность большого числа стадий и, конечно, не предполагала возможности какого-либо практического использования.
Совершенно неожиданно через 10 лет было обнаружено, что родственные соединению 36 метаболиты грибов обладают сильнейшим действием как ингибиторы образования новых кровеносных сосудов (ангиогенеза) и могут быть полезными как противоопухолевые лекарственные средства. Эти данные побудили Кори провести тесты на эту же активность для природного энантиомера (-)-36, в результате которых было установлено, что это соединение более активно и менее токсично, чем изученные ранее метаболиты сходного структурного типа. Этот результат, естественно, заставил снова обратиться к проблеме полного синтеза соединения 36, и ранее описанная процедура была значительно упрощена и приспособлена для получения требуемого энантиомерно чистого (-)-овалицина [22b].1.3. Синтез как поиск (цель бесспорна, но не однозначна)
Синтез природных веществ, в том числе обладающих полезными свойствами, — это лишь одна, наиболее очевидная, но далеко не единственная задача органического синтеза. Как показывает весь опыт органической химии, вещества, обладающие полезными свойствами, могут быть получены не только путем копирования «изделий» Природы. Очень часто практически полезные вещества получаются в результате исследований, совершенно не связанных с синтезом природных соединений. Здесь, однако, возникает серьезная проблема, а именно: какие критерии могут быть положены в основу выбора конкретной цели синтеза и в какой степени предсказуемы свойства ранее неизвестного соединения?
В действительности, многие свойства вещества могут быть надежно предсказаны a priori просто из анализа его структурной формулы. Так, не очень трудно четко указать на некий минимальный набор структурных элементов, которые должны присутствовать в молекуле вещества для того, чтобы оно обладало, скажем, свойствами красителя, душистого или взрывчатого вещества, инсектицида, клея, пластификатора или даже лекарства определенного спектра действия. Предсказания такого рода могут быть сделаны как на основании аналогии с уже имеющимися данными, так и на основе серьезного теоретического анализа. Однако наиболее общим и устойчивым свойством рекомендаций, полученных в результате такого анализа, является их неоднозначность. Покажем сказанное на ряде примеров.
Вероятно, исторически первой областью органической химии, которая быстро и уверенно пошла по пути целенаправленного создания веществ с заранее заданными свойствами, была химия органических красителей. Здесь очень рано были сформулированы эмпирические правила (впоследствии превратившиеся в строгую теорию), связывающие структуру молекул с цветом вещества. В основе этих представлений лежит понятие о хромофоре — группировке атомов, ответственной за характерное для вещества поглощение света определенной длины волны. Одним из распространенных хромофоров синтетических красителей является диарилазогруппа, присутствующая, например, в молекуле азобензола (37, схема 1.12).
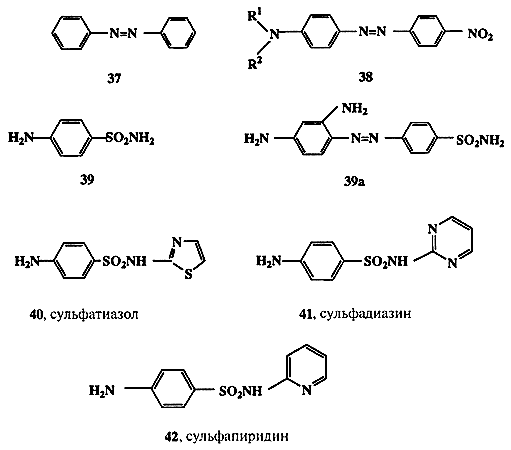
Схема 1.12
Подробно изучено влияние других группировок, связанных с этим хромофором, на спектральные характеристики вещества и, следовательно, на его цвет. Так, родоначальное соединение этого ряда, 37, имеет сравнительно слабую оранжевую окраску, в то время как азосоединения общей формулы 38, содержащие диалкиламино- и нитрогруппы в пара-положениях, окрашены в ярко-красный цвет. Для всех соединений этого ряда, отличающихся лишь вариацией алкильных остатков R1 и R2, можно уверенно предсказать, что для них общими будут красный цвет и слабоосновные свойства (из-за наличия аминогруппы), а также способность изменять цвет в зависимости от рН среды.
Следовательно, если задача будет сформулирована как синтез ярко-красного азокрасителя с основными свойствами, то теория приведет нас к структуре 38, но ничего не скажет о природе алкильных групп. Поэтому исследователь окажется перед необходимостью выбора из неопределенно большого числа близко родственных, но различных соединений. При этом ему придется учесть ряд дополнительных соображений.
Так, от структуры алкильных групп будут зависеть такие характеристики веществ ряда 38, как степень основности, растворимость в воде и органических растворителях, температура плавления, способность к связыванию с поверхностью той или иной ткани, а также термо- и светостойкость. Все эти особенности уже не могут быть а priori предсказаны сколько-нибудь точно. Поэтому в подобных случаях даже после тщательного анализа, как правило, остается несколько почти равноценных структур, и химику придется синтезировать их все. И только лишь после подробного изучения свойств всех этих соединений можно окончательно выбрать те немногие из них, которые отвечают заданным практическим требованиям.
Эмпирический выбор перспективного соединения среди большого числа родственных кандидатов особенно характерен для работ по созданию новых лекарственных и вообще биологически активных препаратов. Здесь теория (а чаще простая эмпирика) позволяет лишь предположить (но отнюдь не гарантировать!), что те или иные соединения, содержащие определенный набор структурных фрагментов, будут проявлять желаемую активность. Множество же других важнейших особенностей будущего лекарства, таких, как, токсичность, способность накапливаться в организме или, наоборот, быстро выводиться из него, возможные краткосрочные или долговременные побочные эффекты, комплекс физико-химических свойств, обусловливающих различные возможности введения в организм и устойчивость при хранении и стерилизации, совместимость с другими лекарственными препаратами и многие другие, почти не поддаются априорной оценке. Поэтому после обнаружения перспективной биологической активности того или иного вещества, выделенного из природного источника или синтезированного в лаборатории, всегда следует серия работ по синтезу ряда его аналогов и сравнительное изучение всего комплекса их свойств, существенных для оценки возможностей их практического использования.
Классическим примером может служить история создания сульфаниламидных препаратов [23]. Первоначальным стимулом к изучению этих производных явилось наблюдение, что наличие сульфаниламидной группировки в молекуле азокрасителя резко увеличивает его способность связываться с шерстяными волокнами. В то время считалось также, что поскольку стенки бактерий состоят в основном из белковых молекул (что совершенно ошибочно!), то сульфаниламиды могут активно связываться и со стенками бактерий и таким образом ингибировать их рост. В ходе дальнейших исследований совершенно случайно была обнаружено, что сульфаниламид 39а, “красный пронтозил” (схема 1.12), обладает удивительно высокой активностью против стрептококковой инфекции на мышах. Напомним, что в те времена (1932 г.) не было эффективных лекарственных средств против бактериальной инфекции. Наиболее удивительной особенностью лекарственного действия 39а было то, что препарат был очень активен в испытаниях на животных in vivo, но его активность падала почти до нуля при стандартных тестах на культурах бактерий in vitro. Разгадка этой тайны была вскоре найдена. Оказалось, что 39а сам по себе неактивен по отношению к стрептококкам, но в организме мыши он претерпевает восстановление (хорошо известная реакция азосоединений), давая сульфаниламид (39), который и является активным бактерицидом.
Вслед за этим открытием последовал настоящий взрыв синтетической активности, и к 1947 г. было получено и испытано в качестве потенциально полезных лекарственных препаратов более 5000 сульфаниламидов. В результате столь широкого скрининга удалось отобрать примерно 100 соединений, обладающих желаемым комплексом свойств. Из них менее дюжины оказались в конце концов практически пригодными в качестве лекарств, а такие, как, например, соединения 39-42 (схема 1.12), до сих пор находятся в арсенале средств борьбы с бактериальными инфекциями.
Понятно, что, если возможно четко установить, какой именно структурный фрагмент ответствен за биологическую активность соединения (как, например, сульфаниламидная группа в соединениях 39-42), поиск соединения, способного стать эффективным лекарственным препаратом, резко ускоряется. Однако лишь в очень редких случаях ситуация столь проста и, как правило, зависимость структура — активность имеет гораздо более сложный характер. В качестве примера рассмотрим некоторые данные о биологической активности стероидных соединений.
Первый стероид, холестерин (43, схема 1.13), был выделен еще в XVIII веке из камней желчного пузыря. Никакой биологической активности этого соединения в то время обнаружено не было. С тех пор еще сотни стероидных соединений были выделены из природных источников, и многие тысячи их аналогов синтезированы в лаборатории. Спрашивается, зачем это надо было делать? Эту проблему проще всего проиллюстрировать, если сравнить структуры некоторых биологически активных соединений этого класса, например 43-50.
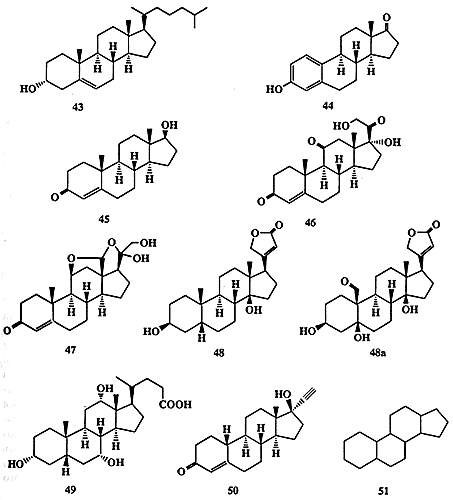
Cхема 1.13Легко заметить близкое структурное родство этих соединений — все они содержат одну и ту же тетрациклическую систему 51, так называемое пергидроциклопентанофенантреновое ядро. Тем не менее, их конкретные функции в живых организмах разительно различаются.
Холестерин (43) присутствует в значительных количествах (его нормальное содержание в организме взрослого человека составляет более 0, 2 кг) практически во всех тканях как составная часть липидной мембраны. Он также пользуется дурной славой из-за своей роли (может быть, не вполне заслуженной) в развитии коронарных заболеваний.
Эстрон (44) и тестостерон (45) являются, соответственно, женским и мужским половыми гормонами млекопитающих.
Кортизон (46) относится к числу гормонов коры надпочечников и обладает, помимо прочего, мощным противовоспалительным действием.
Альдостерон (47) является гормоном, регулирующим солевой обмен, а дигитоксигенин (48) — компонентом дигиталиса, традиционного лекарства «от сердца», выделяемого из некоторых целебных трав, например, наперстянки Digitalis purpurea (отметим, что сходными лекарственными свойствами обладает также близкий по строению строфантидин (48а), рамнозид которого, конваллотоксин, является действующим началом обычного ландыша Convallaria majalis).
К этому списку можно добавить ранее уже упоминавшиеся гормон окукливания насекомых, экдизон (9) и половой аттрактант водоросли 30.
Природные стероидные соединения, как правило, выполняют множество различных функций в организмах. В медицине же обычно желательно иметь лекарство, обладающее некоторым строго определенным набором фармакологических свойств с минимумом побочных эффектов. Однако, даже на примере довольно ограниченной выборки стероидных соединений, представленной на схеме 1.13, легко убедиться в невозможности установления какой-либо однозначной зависимости между их биологической активностью и наличием того или иного структурного фрагмента в молекулах этих веществ.
Так, гидроксильная группа при С-З имеется в соединениях 9, 30, 43, 44, 48, 48а и 49, в то время как 45-47 и 50 содержат при этом центре карбонильную группу. Дополнительные заместители при С-17 имеются в структурах 9, 30, 43 и 46-50. Очевидно, что наличие этих структурных особенностей в молекулах упомянутых выше природных веществ или их синтетических аналогов само по себе не дает возможности предсказать характер их биологического действия. Поэтому единственный реальный способ решения проблемы создания стероидных препаратов с заданным комплексом свойств — это синтез огромного числа аналогов природных соединений и комплексное исследование особенностей их биологического действия.
Синтетические исследования в области стероидов развивались по двум направлениям: полный синтез природных соединений и их аналогов и поиск путей трансформации доступных природных стероидов в практически важные вещества. В результате этих исследований лабораторным путем были получены практически все важнейшие представители этого класса природных соединений, а также их многочисленные аналоги, отвечающие почти всем мыслимым модификациям базовых структур. Среди выдающихся по своей практической значимости достижений в этой области следует упомянуть частичный синтез кортизона (46) из легко доступной холевой кислоты (49). Это превращение первоначально включало 37 стадий и приводило к получению нужного соединения с очень малым общим выходом (Саррет, 1946) [24а]. Трудно было даже предполагать, что этот путь может иметь какое-либо практическое значение. Однако менее чем через 3 года схему синтеза удалось значительно упростить, поднять на несколько порядков суммарный выход в этом превращении и наработать на пилотной установке около 1кг кортизона (46), количества вполне достаточного для проведения клинических испытаний этого важного лекарства [24b].
На протяжении нескольких десятилетий стероиды оставались в числе важнейших целей органического синтеза [24с]. Благодаря интенсивным исследованиям в этой области не только удалось сделать доступными для практического использования в медицине все стероидные гормоны, но и в значительной степени прояснить механизм их биологического действия. В частности, именно результаты таких исследований позволили, наконец, разработать эффективный подход к медицинскому контролю рождаемости — одной из наиболее жгучих проблем современного мира. В настоящее время сотни миллионов женщин во всех странах мира пользуются оральными контрацептивами, созданными на основе соединения 50 (the pill) — синтетического аналога природного гормона прогестерона. Социальные последствия внедрения в практику этого препарата огромны, и, наверное, нелегко подобрать еще один пример того, как появление на планете всего лишь одного нового соединения смогло повлиять на характер жизни значительной части человечества.
О важных результатах других синтетических исследований в области химии стероидов мы еще неоднократно будем говорить в последующих главах нашей книги.
Не менее интересна и история развития синтетических исследований в области простагландинов. Уникальность биологических функций этого класса соединений и крайняя ограниченность природных источников их выделения с необходимостью требовали осуществления полного синтеза этих соединений [например, ПГЕ1 (5, схема 1.2)]. По сути дела успехи медико-биологических исследований, направленных на выяснение функций этих регуляторов, были в первую очередь обусловлены успешным решением задачи их синтеза. Не менее актуальной была задача синтеза разнообразных аналогов этих соединений, что было обусловлено не только крайне низкой стабильностью природных простаноидов, но также тем, что последние выполняют множество самых различных функций в регуляции жизнедеятельности организма. За короткий срок удалось синтезировать несколько сотен стабильных аналогов простагландинов, среди которых и были найдены вещества узко направленного спектра действия, удовлетворяющие требованиям их практического использования [5].
Рассмотрим еще один пример несколько иного плана. Начало истории антималярийных средств датируется серединой XVI в., когда миссионер-иезуит в Перу выяснил, что в качестве эффективного средства борьбы с малярией индейцы используют кору хинного дерева. В 1834 г. Пеллетье выделил активное начало этого средства, хинин (52, схема 1.14), но его строение было установлено лишь в XX в.
Интересно, что первая попытка синтеза хинина была предпринята 18-летним Уильямом Перкиным (впоследствии ставшим знаменитым химиком-синтетиком) в 1856 г., т.е. во времена, когда само представление о том, что молекула может иметь какое-то определенное строение, собственно еще только начало складываться в сознании химиков. Зная только брутто-формулу хинина, Перкин предположил, что окисление смеси толуидина и аллилбромида азотной кислотой даст хинин. Эта дерзкая попытка, конечно, провалилась, но в результате реакции вместо хинина Перкин получил первый синтетический краситель, мовеин.
В дальнейшем предпринимались более осмысленные попытки полного синтеза хинина, увенчавшиеся успехом лишь в 1948 г. Параллельно этим работам также проводились поиски более дешевых средств против малярии. Были получены и испытаны тысячи возможных кандидатов на эту роль, иногда имеющих более чем отдаленное сходство с хинином. Некоторые из них, такие, как хлорохин (53), мепакрин (54) или прогуанил (55) (схема 1.14), нашли широкое применение в медицине как эффективные заменители хинина [25а].
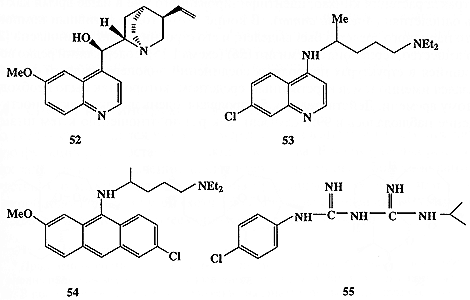
Схема 1.14
Могло показаться, что тем самым проблема борьбы с малярией была окончательно решена. Однако, как это часто бывает в химиотерапии, широкое использование противомалярийных средств привело к появлению мутировавшего штамма Plasmodiurn falciparum — паразита, вызывающего малярию, — и этот новый штамм оказался резистентным к этим лекарственным препаратам. Возникла проблема создания препаратов нового поколения и снова, как это ни парадоксально для второй половины XX в., решение помогли найти средневековые рецепты народной медицины, на этот раз китайской.
Еще в 1596 г. известный китайский целитель-травник Ли Шичен в трактате “Compendium Materia Medica” описал использование для лечения малярийной лихорадки экстракта растения кинхао, идентифицированного уже в наше время как полынь однолетняя (Artemisia аnnua). В конце 1970-х годов активный компонент этого народного средства был выделен, и его строение было установлено [25b]. Это вещество, терпеноид артемизин (56) (схема 1.15), содержащий редко встречающийся в структурах природных соединений эндопероксидный фрагмент, оказался именно тем лекарственным средством, которое так было необходимо к этому времени. Действительно, 56 проявлял очень высокую активность (излечение наблюдалось в 99% случаев) как раз по отношению к тем штаммам P. falciparum, которые были резистентными по отношению к хлорохину (53); более того, он оказывался эффективным даже против церебральной малярии — поздней и обычно летальной фазы заболевания.
Однако практическое применение соединения 56 было затруднено, поскольку это вещество мало растворимо в воде, химически неустойчиво и к тому же довольно дорого. На помощь вновь пришел органический синтез. Полный синтез артемизина не был уж очень сложной задачей, но тем не менее реализованные к настоящему моменту схемы его получения (см., например, ссылки в [25с]) вряд ли имеют перспективы практического использования. Однако благодаря интенсивным синтетическим исследованиям в этой области удалось не только отработать эффективные методы создания эндопероксидного фрагмента, ключевого элемента структуры 56, но и получить достаточно представительный набор его аналогов. Первичные испытания показали, что некоторые из стабильных аналогов, такие, как, например, соединения 57 и 58, даже более активны в тестах in vitro, чем 56 [25d].
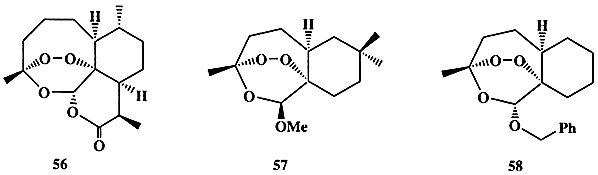
Схема 1.15
Большая часть синтетических исследований, упомянутых в настоящем разделе, обычно классифицируются как “синтез потенциально полезных веществ”. В проблемах этого типа, не имеющих однозначного ответа, химик приучается мыслить не только в терминах единичных целевых структур, а скорее категориями больших серий родственных соединений. Поэтому особое значение приобретает разработка достаточно общих синтетических схем, допускающих получение наборов соединений, отличающихся закономерными вариациями структурных параметров. Именно наличие таких наборов служит основой для установления корреляций “структура — свойство”, без чего, конечно, невозможно вести рациональный поиск оптимальной (для данной цели) структуры. Иными словами, это стиль систематического поиска, трудоемкий, иногда даже скучный, но пока неизбежный. И хотя нередко из тысячи синтезированных разными исследователями соединений “в дело” пойдет лишь одно, нельзя считать, что 999 других были получены напрасно: ведь это одно было выбрано из всей тысячи *! Без всего массива синтезированных и изученных соединений эта оптимальная структура вряд ли была найдена, ибо вероятность ее случайного обнаружения не превышает 1 : 1000.
* Приведенное соотношение не является просто гиперболой. Соотношение 1 : 1000 считается вполне приемлемым в работах, направленных на создание биологически активных соединений. В последнее десятилетие это положение начинает меняться в лучшую сторону благодаря разработке принципов молекулярного дизайна, о чем пойдет речь в главе 4.1.4. Синтез как инструмент исследованияВо всех обсуждавшихся выше примерах синтез выполняет чисто препаратив-ную функцию, т.е. поставляет нужные вещества. В принципе для решения таких задач не имеет значения, каким именно путем было получено данное вещество: его можно было выделить из природных источников или получить микробиологическим путем (хотя в большинстве случаев именно химический синтез оказывается наиболее общим и надежным способом).
Однако в некоторых областях синтез используется не просто в качестве удобного “подручного средства”, а составляет самую суть задачи. Речь идет, в первую очередь, о встречном синтезе природных соединений или соединений, впервые полученных в результате неизвестных ранее химических превращений. В таких случаях наиболее надежным и бесспорным доказательством справедливости структуры, выведенной на основании обычных методов анализа, является его химический синтез и установление идентичности полученного вещества заведомого строения с исследуемым веществом.
Подобное утверждение сегодня может показаться устаревшим. Действительно, такие мощные современные методы структурного анализа, как спектроскопия в ультрафиолетовой, видимой и инфракрасной областях, ядерный магнитный резонанс, масс-спектрометрия высокого разрешения, казалось бы, обеспечивают возможность быстрого установления структур любой сложности. Однако нельзя забывать, что трактовка результатов, получаемых любым из этих физических методов, базируется прежде всего на аналогиях с известными соединениями и потому тем более надежна, чем ближе структура изучаемого соединения к уже изученным. В случае же необычных, новых типов структур интерпретация спектров в структурных терминах может стать далеко не тривиальной задачей. Конечно, существует метод, свободный от этих ограничений, а именно рентгеноструктурный анализ (РСА). Этот метод справедливо считается абсолютным методом установления структуры, однако ему присущи ограничения другого, технического характера — он применим лишь в тех случаях, когда можно получить монокристалл исследуемого вещества, что достижимо далеко не всегда.
Другой, классический подход к установлению структуры вещества основан на его деструкции, т.е. на химической “разборке” молекулы на составные части и логической реконструкции исходной структуры на основе информации о строении этих составных частей. Этот путь, несмотря на его универсальную применимость, очень трудоемок и предполагает наличие значительных количеств анализируемого вещества. К тому же, в ходе подобной деструкции почти неизбежна потеря части информации (в особенности стереохимической) относительно строения тех участков молекулы, по которым производится ее деструкция.
От всех упомянутых выше ограничений свободен встречный синтез. Если структура исследуемого вещества подтверждена его встречным синтезом, то она может считаться полностью доказанной, и этот “вердикт” не подлежит пересмотру.
Нередки ситуации, когда встречный синтез вообще оказывается единственным средством выбора между несколькими альтернативными структурами изучаемого вещества. Так бывает в тех случаях, когда вещество доступно в ничтожно малых количествах — в долях миллиграмма или даже микрограммах, — которых явно мало для использования деструктивных методов анализа и даже для применения современных спектральных методов. В то же время этих количеств вполне может хватить для идентификации вещества, т.е. установления тождественности двух его образцов, например, синтетического и природного.
Именно встречный синтез позволил окончательно установить структуры таких природных веществ, как уже упоминавшиеся ювенильный гормон (8) или бомбикол (12). Ниже мы рассмотрим еще несколько более недавних примеров.
В 1976-78 гг. ценой напряженного труда удалось выделить в чистом виде один из активнейших половых феромонов самки таракана Periplaneta ameriсаnа. Активность выделенного вещества, перипланона В, оказалась поистине удивительной — порог чувствительности особи составлял всего 10-13 г, и оно рассматривалось как перспективное средство борьбы с этим вездесущим насекомым. Однако в распоряжении исследователей имелось лишь ничтожно малое количество этого вещества (около 200 мкг перипланона В, выделенные из 75000 особей). Даже использование всего арсенала средств инструментального анализа не позволило однозначно установить его строение. С помощью этих методов удалось лишь выяснить некоторые основные особенности структуры, а именно: строение углеродного скелета, характер и распределение функциональных групп, как это показано в формуле 59а (схема 1.16), но вопрос о стереохимии молекулы оставался открытым. Эту самую сложную часть задачи удалось решить лишь после того, как был выполнен полный синтез трех из четырех возможных диастереомеров перипланона В [26а]. Прямое сравнение синтезированных образцов с природным позволило установить, что стереохимия последнего соответствует показанной в формуле 59b. Минорный компонент природного феромона, перипланон А, был выделен в еще меньших количествах, и по данным первоначальных спектральных исследований ему была ошибочно приписана структура 60а. Истинную структуру 60b удалось выяснить только путем полного синтеза этого соединения [26b]. Благодаря интенсивным синтетическим исследованиям в этой области, был получен также ряд биологически активных аналогов 59а (как, например, 61) из достаточно доступных предшественников [26с].
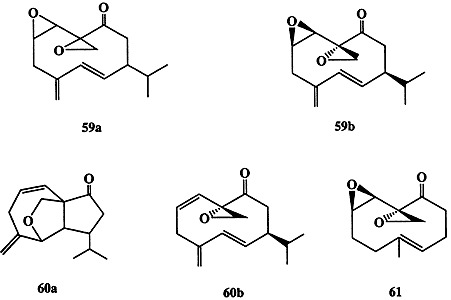
Схема 1.16
В 1990 г. японским химикам удалось выделить из культуральной жидкости микроорганизма Streptoverticillium fervens новый антигрибковый агент, обозначенный как FR-900848, для которого была установлена общая структура 62а (схема 1.17). Весьма интересной оказалась активность этого агента против нитевых грибов, особенно опасной инфекции для больных СПИДом или диабетом [27]. В течение ряда лет стереохимию этого соединения не удавалось установить, так как даже самые изощренные методики ЯМР-спектроскопии оказались бессильными для решения задачи установления стереохимии для полициклопропанового остова 62а, содержащего 10 хиральных центров. Было также очевидно, что в этом случае вряд ли разумно было пытаться решить эту проблему путем синтеза всех (210!)диастереомеров этого соединения. Однако рассмотрение возможных путей биосинтеза этого соединения в живой клетке позволило предположить, что наиболее вероятной является полностью транс-конфигурация(all-trans) заместителей во всех циклопропановых остатках [27а].
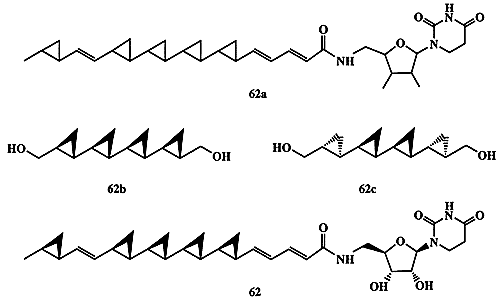
Схема 1.17
При частичной деградации природного продукта было получено симметричное производное кватерциклопропана, которому можно было приписать структуру 62b или 62с. Оба этих диастереомера были получены независимым синтезом с использованием стереоспецифичных реакций, что позволило однозначно установить полностью транс-, полностью син-конфигурацию (all-trans, all-sin) продукта деградации, т.е. структуру 62b. Этот результат, в сочетании с рядом других данных, позволил установить полную структуру FR-900848, показанную в формуле 62 [27b]. Попутно заметим, что вскоре был выделен следующий представитель полициклопропанов из культуральной жидкости ряда видов стрептомицетов Streptomyces spp. Это соединение, проявляющее значительную активность как ингибитор отложения липидов на стенках артерий, отличается от соединения 62 наличием еще одного циклопропанового звена и, по-видимому, принадлежит к тому же стереохимическому ряду [27с].
Дополнительные сложности при установлении строения могут возникать из-за повышенной лабильности некоторых природных соединений. В 1969 г. в результате тщательных биомедицинских исследований было обнаружено, что в тромбоцитах продуцируется, хотя и в очень малых количествах, вещество, являющееся чрезвычайно мощным фактором сужения сосудов и агрегации тромбоцитов. Имелись довольно серьезные основания полагать, что результатом “перепроизводства” этого фактора, тромбоксана А2 (ТхА2), может быть сердечный приступ или инсульт.
Выделить этот фактор удалось лишь в 1975 г., но его крайняя нестабильность (период полураспада этого вещества в водном растворе при 37°С составляет всего 34 с!), если и не исключала, то, по крайней мере, крайне ограничивала возможность использования для установления его строения стандартных физико-химических методов. Однако не составило большого труда выяснить, что образующееся в результате разложения тромбоксана биологически инертное вещество имеет строение 6За (схема 1.18) [28а].
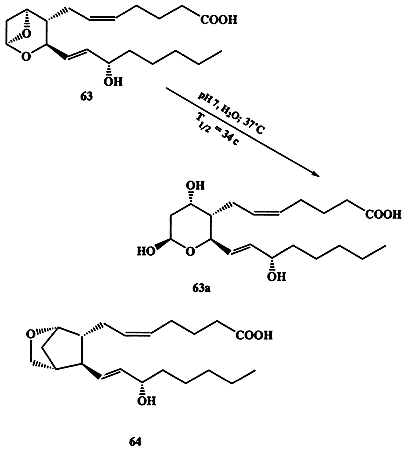
Cхема 1.18
Этот факт в сочетании с данными, полученными при изучении путей биогенеза всего семейства простаноидов, позволил предложить структуру 63 как наиболее вероятную для тромбоксана А2. Эта структура казалась настолько необычной для природного соединения, что на протяжении почти 10 лет она неоднократно оспаривалась. Тем не менее, именно данная структура была однозначно подтверждена в 1985 г., когда в лаборатории Стилла [28b] был выполнен полный синтез ТхА2. Интересно, что в данном случае только один единственный критерий мог быть использован для доказательства идентичности образцов природного и синтезированного веществ, а именно тождественность их биологической активности в серии стандартных тестов. Исключительная нестабильность соединения 63 оказалась также серьезным препятствием для экспериментального изучения его физиологических функций, что побудило предпринять серию исследований, направленных на синтез химически стабильных аналогов 63. Среди множества последних одним из наиболее эффективных заменителей 63 оказалось соединение 64 [28с].
Однако помимо той, иногда действительно ключевой роли, которую играет синтез при решении задач установления строения, у него есть еще другая, менее очевидная, но более общая и, пожалуй, более важная функция. Дело в том, что синтетическое исследование само по себе является наиболее мощным инструментом активного познания химии синтезируемых соединений. Действительно, только глубокое понимание химического своеобразия органического соединения позволяет успешно осуществить его целенаправленный синтез, но сами знания, необходимые для подобного понимания, эффективно накапливаются именно в ходе выполнения этого синтеза. Почему это так?
Первоначальный план синтеза обычно строится на хорошо известных синтетических методах, принципиальная пригодность которых для решения конкретной задачи не вызывает особых сомнений. И если все идет по плану, то мы тем самым получаем экспериментальное подтверждение справедливости наших предсказаний о химии конкретных соединений, участвующих в предпринятом синтезе. По этому поводу один из величайших синтетиков XX века лауреат Нобелевской премии Р. Вудворд писал : «Вряд ли можно отрицать, что успешный исход синтеза, состоящего из более чем 30 стадий, является суровым испытанием способности науки к предвидению, а также проверкой ее познавательной мощи в сфере изучаемых объектов» [29].
Однако нередко при вторжении в новую «сферу изучаемых объектов» хорошо апробированные методы не срабатывают, и здесь и начинается самое интересное.
Во-первых, сам факт такой «осечки» — это уже небольшое (а иногда и значительное) открытие — обнаружение неожиданной химической особенности, присущей изучаемой структуре. Причем маловероятно, чтобы эта особенность была обнаружена “просто” при изучении химии данного соединения вне связи с его синтезом. Маловероятно именно потому, что речь идет о хорошо изученных реакциях, результат которых казался точно предсказуемым и поэтому, если бы не потребности синтеза, то вряд ли имело смысл ставить такой эксперимент.
Во-вторых, существование конкретной синтетической цели не позволяет исследователю ограничиться индифферентной констатацией факта, что такая-то реакция в данном случае странным образом не идет или идет, но не так, как предполагалось. Затруднение нужно преодолеть, и наиболее эффективный путь для этого — понять причину возникновения аномалий, т. е. глубже разобраться в химии изучаемых соединений. Понимание причин наблюдаемых осложнений может помочь в разработке нового варианта примененного метода. Если и таким путем преодолеть препятствие не удается, то приходится искать какие-то новые пути и привлекать для этого и методы и идеи из других, часто отдаленных областей химии. Такой напряженный и многогранный целенаправленный поиск решения дает в качестве “побочного продукта” новые и углубленные знания о реакционной способности органических соединений — знания, которые вряд ли бы были получены в обозримое время, если бы не возникло проблем с решением поставленной синтетической задачи.
В справедливости сказанного читатель легко может убедиться, обратившись к серии блестящих синтезов, выполненных в группе Кори. В своей Нобелевской лекции Кори специально подчеркнул, что “ключом к успеху множества многостадийных синтезов, которые были осуществлены в нашей лаборатории за последние годы, было изобретение новой методологии” [30]. Согласно его оценкам, не менее 50 новых методов было создано в ходе этих исследований.
Более того, подчас именно благодаря “осечкам” при применении хорошо известных методов к решению конкретной и казалось бы частной синтетической задачи удавалось обнаружить принципиально новые особенности строения и реакционной способности исследуемых объектов. Их последующее изучение приводило к результатам, важным для всей органической химии, далеко за пределами “сферы изучаемых объектов”. Поясним это утверждение некоторыми примерами.
В 1900 г. Гомбергом проводилось исследование, целью которого был синтез гексафенилэтана (65). По тем временам эта структура казалась довольно экзотичной, но на самом деле тем же Гомбергом ранее (в 1897 г.) уже был синтезирован, хотя и с незначительным выходом, тетрафенилметан, и, вообще говоря, не было оснований ожидать особых осложнений при синтезе 65, “всего лишь” следующего представителя перфенилированных производных алканов. Однако использование стандартного в то время метода создания углерод-углеродной связи — конденсации галогенпроизводных в присутствии металлов (схема 1.19) — привело к более чем странным результатам: вообще не наблюдалось образования углеводорода 65 при реакции трифенилхлорметана с каким-либо из металлом (Zn, Ag или Hg) в обычных условиях проведения подобного рода конденсаций. При этом в качестве единственного продукта реакции был получен пероксид 65а [31а].
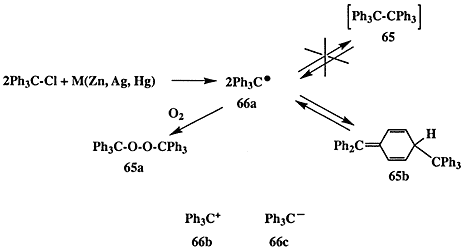
Схема 1.19
Естественно, что следующим шагом было проведение реакции в условиях, исключающих наличие кислорода воздуха (в атмосфере CO2), и в этом случае удалось получить углеводород, отвечающий по составу гексафенилэтану (65). Однако совершенно неожиданно было обнаружено, что полученный углеводород ведет себя в растворе как димер, способный к диссоциации в растворе на две идентичные субъединицы, и в присутствии кислорода он быстро превращается в тот же пероксид 65а.
Анализ этих и других экспериментальных данных привел Гомберга к выводу о том, что на самом деле непосредственным результатом реакции трифенилхлорметана с металлом является образование трифенилметила как стабильной частицы 66а [31а, b], которая в растворе может претерпевать обратимую димеризацию в гексафенилэтан (65). В то время требовалась немалая смелость для подобного заявления, подразумевавшего возможность существования соединений, в которых углерод трехвалентен, поскольку господствовало убеждение, что углерод бывает либо четырехвалентным, либо — в редких случаях — двухвалентным (например, в монооксиде углерода или изонитрилах, R-NC). Поэтому вполне естественно, что результаты синтетических опытов Гомберга были встречены с большим сомнением.
Вскоре, однако, достоверность этих результатов была строго подтверждена и стало ясно что итогом «синтетической неудачи» Гомберга * явилось открытие свободных радикалов — нового класса химических частиц, производных трехвалентного углерода. Дальнейшие исследования показали, что для такой же трифенилметильной системы могут быть получены и производные трехвалентного углерода других типов, а именно соли трифенилметил-катиона (66b) и трифенилметил-аниона (66с).
* Отметим, что на протяжении более чем полувека так и оставался нерешенным вопрос о строении димерного углеводорода, которому первоначально было приписано строение гексафенилэтана. Лишь в 1968 г. было строго доказано (с помощью метода ЯМР), что полученный Гомбергом углеводород на самом деле имеет хиноидную структуру 65b [31 с]. «Настоящий» гексафенилэтан (65) так и не получен до сих пор [31d].Получение 66а-с, первых представителей соединений нового структурного типа как стабильных веществ, а также выяснение факторов, обусловливающих их стабильность и особенности реакционной способности, послужило мощным импульсом для разработки новых представлений о механизмах органических реакций, которые предусматривали промежуточное образование подобного рода интермедиатов. Возникновение этих представлений сыграло ключевую роль в становлении классической теории органической химии.Подчеркнем еще раз, что первичным толчком, “запустившим” всю серию столь важных по последствиям исследований, явилась работа Гомберга, преследовавшая сугубо препаративные цели.
Не менее выразительный пример первостепенной роли синтеза в развитии теоретических представлений может быть найден в истории полного и частичного синтез стероидных гормонов и их аналогов. Исследователи, работавшие в этой области в 1930—40-х годах, встретились с рядом неожиданных проблем как при построении углеродного скелета, так и при осуществлении некоторых иногда вполне тривиальных превращений, таких, как присоединение по связи С=С или С=O, раскрытие оксиранового цикла или даже превращение спиртов в соответствующие галогенопроизводные. Потребности синтеза не только заставили химиков разработать альтернативные методы, позволявшие осуществлять такие превращения, но и побудили обратиться к изучению причин наблюдаемых аномалий.
Именно благодаря глубокому анализу особенностей реакционной способности функциональных групп в конформационно закрепленных системах (а к таким системам относится тетрациклический остов стероидов) и удалось сформулировать основные понятия современного конформационного анализа. Напомним, что еще в 1890 г. Заксе [32а] предположил, что циклогексан не является плоской молекулой и сделал вывод о том, что “все монозамещенные производные циклогексана могут существовать по крайней мере в виде двух модификаций”.
Поскольку в то время не имелось никаких экспериментальных данных в пользу этого, вообще говоря, вполне разумного предположения (вспомним, хотя бы тот факт, что к этому моменту тетраэдрическая модель атома углерода Вант-Гоффа и Ле Беля уже была общепринятой), о нем никто особенно и не вспоминал в последующие 60 лет, хотя за это время появился ряд теоретических и физико-химических исследований, свидетельствовавших о правомерности подобного рассмотрения. Но в полной мере прозорливость предположения Заксе могла быть оценена лишь после публикации в 1950 г. в журнале Experientia короткого сообщения под названием “Conformation of the Steroid Nucleus” [32b]. Автор этой работы Бартон проделал огромную работу по сбору и обобщению многочисленных и часто противоречивых данных по реакционной способности различных замещенныхстероидов и собрал убедительные доказательства того, что наблюдаемые различия обусловлены прежде всего значительным различием свойств заместителей в зависимости от того, находятся они в аксиальном или экваториальном положениях, а также особенностями стереохимии всей молекулы. На основе этих представлений вскоре была развита вся концепция конформационного анализа [32с], и пионерский вклад Бартона в ее создание был справедливо отмечен Нобелевской премией 1969 г.
В настоящее время представления конформационного анализа стали неотъемлемой частью теоретических основ органической химии. Трудно представить себе, что на протяжении почти 100 лет этой концепции вообще не существовало и что ее рождение было обусловлено более всего неотложными потребностями органического синтеза.
Органический синтез сегодняшнего дня успешно решает задачи получения молекулярных конструкций невероятной сложности. Закономерно, что на каждом этапе усложнения целей органического синтеза возникают неожиданные синтетические проблемы, обнаруживаются специфические особенности строения и реакционной способности, что не только стимулирует разработку новых синтетических методов, но и служит постоянным источником фактического материала для углубления уже сложившихся и создания новых концепций теоретической органической химии. Поэтому легко представить, насколько беднее и приземленное выглядела бы органическая химия, если бы по какой-либо причине ей пришлось отказаться от синтеза как одной из главных своих задач *.
* Это предположение не столь гипотетично, как может показаться читателю, особенно молодому. Достаточно напомнить, что в 60-х годах усилиями некоторых ученых-чиновников Академии наук СССР работы по полному синтезу были отнесены к разряду второстепенных, что безусловно и явилось одной из причин все нарастающего отставания отечественной органической химии от мирового уровня.1.5. “Химия создает свой предмет...”Еще в 1860 г. выдающийся химик XIX в. М. Бертло писал: “Химия создает свой предмет. Эта творческая способность, подобная искусству, коренным образом отличает химию от остальных естественных и гуманитарных наук” [33]. Попробуем разобраться, на чем основывалось подобное представление об исключительном положении химии в ряду других наук.
Действительно, во все времена естествознание занималось изучением Природы, поисками внутренней связи явлений и законов, управляющих этими явлениями. Природа для ученого всегда являлась изначальной данностью, которую надо было исследовать. Так, биолог изучает живую природу в том виде, в каком она сформировалась в условиях Земли. Астроном изучает уже существующие планеты, звезды, галактики и, наконец, всю Вселенную как целое. Объект исследования химика-органика — органические соединения, их свойства, реакции и закономерности поведения. Однако в отличие от своих коллег-естественников химик должен был сначала создать свой объект исследования, причем создать в самом прямом и точном смысле слова, т.е. синтезировать вещества, которые в Природе (или по крайней мере на Земле) никогда не существовали. В этом смысле органическая химия действительно принципиально отличается от всех других естественных наук, и с самого своего начала она составляет систему, которая черпает в самой себе как объекты исследования, так и проблемы, требующие решения, и развивается по своим внутренним законам. Аналогию такой способности к саморазвитию можно найти разве что в математике.
Так обстояло дело во времена Бертло, и в значительной мере так оно обстоит и сейчас, хотя исключительность органической химии в смысле создания своего объекта исследования несколько поколебалась с появлением совсем новых областей науки, таких, как физика твердого тела, нелинейная оптика, генная инженерия и т.д., развитие которых в значительной мере основано на создании сложных искусственных объектов.
Тем не менее основная мысль Бертло остается справедливой и сегодня: органическая химия выступает в роли подлинного творца, постоянно создавая ту самую искусственную природу, которую сама же и исследует, развивает и находит ей области применения.
Более того, и это особенно интересно, свойства этой искусственной природы оказываются столь же разнообразными, неожиданными и неисчерпаемыми, как свойства “обычной” природы. Это и составляет принципиальное отличие синтетических органических соединений от других классов искусственных объектов. В самом деле механические, электрические или логические структуры, создаваемые человеком, могут быть беспрецедентно сложными, не имеющими прототипов в Природе. Но при всей их сложности никаких качественно новых свойств, которые не могли быть предположены на стадии проекта, в них обнаружиться не может, поскольку они проектировались и создавались для совершенно определенных целей.
Например, если мы проектируем и строим самолет, то он может быть лишь хорошим или плохим самолетом, но ни при каких обстоятельствах не окажется вдруг магнитофоном или мясорубкой. Напротив, если мы синтезируем новое соединение, предназначенное служить лекарством, то, вообще говоря, нет никакой гарантии, что оно не окажется токсином, дефолиантом, фотосенсибилизатором или еще чем-то совершенно непредвиденным. Столь же неожиданными могут оказаться результаты синтетических исследований, не преследующих каких-либо прикладных целей.
Так, в середине 1880-х годов молодой русский химик Зелинский, работавший в лаборатории Майера в Германии, разрабатывал новую схему получения тетрагидротиофена (67) из 2-хлорэтанола (68) с помощью подкупающе простой последовательности реакций, показанной на схеме 1.20. Однако осуществление синтеза пришлось внезапно остановить на стадии получения ключевого полупродукта, а именно b,b'-дихлордиэтилсульфида (69).
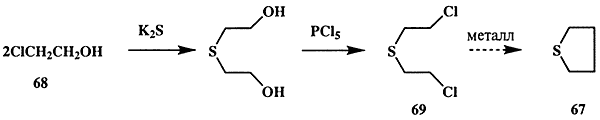
Схема 1.20
Вместо того, чтобы попробовать осуществить последнюю стадию синтеза, внутримолекулярную циклизацию по реакции Вюрца (кстати, в настоящее время можно с уверенностью утверждать, что при обработке 69 металлом ничего, кроме элиминирования не могло произойти), Зелинскому пришлось провести несколько недель в больнице из-за серьезнейших ожогов, вызванных контактом с таким простым и вполне невинно выглядевшим (на бумаге!) соединением, которое позднее приобрело вполне заслуженную дурную славу под названием “иприт”. Однако подобной “зловредной” физиологической активностью вовсе не исчерпываются свойства иприта, и его открытие принесло человечеству не только бедствия. Детальное исследование механизма его действия, вызванное суровой необходимостью в ходе первой мировой войны, привело к созданию нового и по тем временам наиболее эффективного направления в лечении злокачественных опухолей, основанного на использовании иприта и его структурных аналогов в качестве химиотерапевтических средств.
Рассмотренный пример (а число таких примеров исчисляется сотнями!) наглядно показывает, что органические соединения, созданные руками человека, в такой же мере могут служить источником совершенно неожиданных открытий, как и нерукотворные объекты исследований, поставляемых Природой.
Причины такого своеобразия органической химии лежат прежде всего в безграничности числа возможных органических соединений, а следовательно, в безграничном многообразии их свойств (частным проявлением этого многообразия является сам факт существования жизни на Земле).
Общеизвестно, что уникальность углерода состоит в сочетании двух свойств: его четырехвалентности и способности образовывать прочные связи как с другими атомами углерода, так и с атомами многих других элементов. Именно поэтому число возможных органических соединений оказывается бесконечно большим, в строгом смысле этого слова.
Как формулируется понятие бесконечности в математике, скажем, в простейшем случае бесконечности натурального ряда чисел? К любому сколь угодно большому числу можно прибавить единицу и получить следующий член этого ряда, с которым можно проделать ту же операцию, и т. д. Аналогично к любой сколь угодно сложной органической структуре можно присоединить (по крайней мере, теоретически), например, метильную группу и получить новое соединение. С той только разницей, что такую операцию со сложной органической молекулой можно выполнить множеством различных способов, а присоединять можно отнюдь не только метильную группу. Множественность вариантов усложнения проявляется на самых ранних этапах, начиная с молекул, содержащих всего несколько атомов, и число таких вариантов возрастает примерно пропорционально числу уже имеющихся в молекуле атомов углерода.
В таком «ветвящемся дереве» множества структур коэффициент ветвления будет монотонно возрастать с ростом уже пройденных точек ветвления. Общее число членов такой системы должно расти по закону, близкому к факториалу (n!, где n — число атомов углерода в молекуле). Это означает, что даже в пределах не очень больших органических молекул число возможных структур становится поистине астрономическим.
Рассмотрим, например, соединения состава C60 (схема 1.21), относящиеся к узкому классу — насыщенным алифатическим кислотам общей формулы 70, в которых заместителями R1 и R2 в любом возможном положении могут быть любые из десяти показанных на схеме групп.
Схема 1.21
Число подобных структур, возникающих просто при вариации природы и положения всего лишь одной из групп, R1 или R2 , составит 1029. Если варьировать обе группы, то общее число возможных комбинаций составит 1029х1029, что примерно в 107 раз превышает число всех атомов Земли. Всего углерода, имеющегося в нашей Галактике, не хватит на то, чтобы получить все соединения из этого набора даже в миллиграммовых количествах. Каждый из третичных атомов углерода в соединениях 70 является асимметрическим центром, и поэтому любое из них может быть представлено 229 стереоизомерами, что увеличивает общее число структур типа 70 примерно до 5, 4х1066. Для их синтеза (по 1 мг каждого) не хватит уже всех нуклонов во всей наблюдаемой Вселенной. Так, от абстрактной математической бесконечности мы приходим к вполне реальному, поистине неисчерпаемому многообразию органических соединений.
Каковы же источники всего этого многообразия? Как бледная схема теоретически возможных виртуальных структур расцвечивается полнокровными красками реально существующих веществ? Таких источников два: природный (ископаемое органическое сырье и современные живые организмы), и искусственный (органический синтез).
Органическая химия зародилась как химия соединений, выделяемых из живых организмов (чему она и обязана своим названием). Однако природные соединения несмотря на огромное разнообразие их структур заполняют систему органических соединений очень прихотливым и — с чисто органохимической точки зрения — случайным образом, поскольку пути биосинтеза определяются прежде всего биологической целесообразностью, а вовсе не потребностями химической систематики.
Если взять любую рациональную классификацию органических соединений, например, по функциональным группам, и заполнить ее только структурами природных соединений, то мы увидим очень странную картину: отдельные кластеры, густо усеянные разнообразными структурами, области, содержащие лишь отдельные точки, и, наконец, огромные пустые области. В такой системе, например, будут щедро представлены неразветвленные алифатические кислоты с четным числом атомов углерода, но будет мало разветвленных кислот или кислот с нечетным числом атомов углерода; будет множество очень причудливо устроенных циклических и полициклических систем, но почти не встретится их простейших представителей. Редкими и “случайными” структурами будут представлены такие важнейшие классы, как алкилгалогениды, тиолы и сульфиды, нитро- и диазосоединения. Удивительно, но будут отсутствовать даже такие тривиальные соединения, как формальдегид, хлороформ, диэтиловый эфир или тетрагидрофуран. Мы уже не говорим о том, что многие важнейшие классы органических соединений, такие, как, например, различные типы металлоорганических соединений или борорганические производные, вообще никак не представлены в списке природных веществ.
Совершенно ясно, что с таким материалом, каким разнообразным бы он ни был, органическую химию, как науку, создать было бы невозможно. Именно поэтому с первых же своих самостоятельных шагов химики-органики с поразительной смелостью пошли по пути создания своего объекта исследования, синтезируя тысячи и тысячи неизвестных Природе веществ и изучая их свойства и взаимопревращения. Огромные усилия нескольких поколений ученых были потрачены на то, чтобы создать прежде всего фундамент фактов для новой науки — органической химии — и определить проблемы, которыми она должна заниматься. Без этого не могло бы состояться создание грандиозной области науки и промышленности и в конечном счете новой, искусственной природы. Эта созданная руками человека природа не только обеспечивает нас почти всем необходимым для повседневной жизни, но и становится на наших глазах все более значимым биогеохимическим фактором глобального масштаба *.
* Значимость этого фактора состоит не только в общеизвестных проблемах, связанных с загрязнением окружающей среды вредными веществами. Не меньшее значение имеет сам факт появления на нашей планете многочисленных искусственно созданных веществ, что может абсолютно непредсказуемым образом сказаться на эволюции биосферы. Частным примером, иллюстрирующим возможную роль этого нового антропогенного фактора естественного отбора, может служить хорошо известный феномен ускоренного формирования новых резистентных штаммов микроорганизмов, которое индуцируется появлением все новых и новых лекарственных средств.Здесь, конечно, не место излагать историю органической химии, но стоит, хотя бы схематически, проследить те главные линии ее развития, в которых определяющую роль играл и продолжает играть органический синтез.1.5.1. Выяснение закономерностей, связывающих строение соединений с их свойствами
Пожалуй, главная, наиболее фундаментальная задача не только органической химии, но и всей химической науки — это установление зависимости свойств исщества (физических, химических, биологических) как функции главного в химии аргумента — молекулярной структуры. Подобные функциональные зависимости в принципе невозможно установить на примере одного соединения. Чтобы изучить или хотя бы обнаружить функциональную зависимость, надо проварьировать аргумент, т.е. изучить серию соединений различной структуры. Изменения структуры органического соединения могут происходить только дискретно, скачками, и какими бы минимальными они ни были, они в той или иной мере сказываются на всем комплексе свойств вещества. Поэтому любое органическое соединение представляет собой неповторимую химическую индивидуальность с единственной конкретной структурой и единственным набором свойств. Именно поэтому закономерности типа "структура — свойство” могут быть выражены в количественном виде лишь для ограниченного круга задач и объектов (как, например, это удается сделать в гамметовских корреляциях свободной энергии или в рассмотренном выше случае оценки зависимости цветности азокрасителей от природы хромофоров). В большинстве же случаев эти закономерности носят чисто качественный характер, и в поиске вещества с заданными свойствами неизбежен эмпирический подход, который предполагает синтез и всестороннее исследование серий родственных соединений с планомерно варьируемыми свойствами *.
* Нас могут упрекнуть в том, что мы умалчиваем о новых подходах, появившихся благодаря разработке самых разнообразных и эффективных компьютерных программ для решения задач типа “структура— свойство”. Безусловно, использование этих программ может существенно сократить область эмпирического поиска и тем самым ускорить решение таких задач. Хотелось бы, однако, отметить, что эффективность этого подхода в конечном счете более всего зависит от полноты и представительности обрабатываемого банка данных, получаемых все тем же “старым добрым” способом, т.е. путем эксперимента.1.5.2. Создание новых структур, проблемных для органической химииНа протяжении всей истории органической химии в ней возникали и продолжают возникать проблемы теоретического характера, для решения которых необходимо было изучить те или иные соединения с экзотической (по крайней мере, для своего времени!) структурой. Для этого нужно было прежде всего их синтезировать и тем самым убедиться в возможности их существования. Изучение свойств таких соединений не только позволяло проверить справедливость предсказаний теории, но и во многих случаях служило импульсом к созданию новых теоретических концепций. Рассмотренные ниже примеры могут служить иллюстрацией сказанного.
Отдельные случаи изомерии органических соединений были известны уже в начале 1830-х годов, но рациональная интерпретация этого феномена стала возможной лишь позднее, в 1850-х годах с появлением теории строения органических соединений. Действительно, согласно представлениям этой теории, развитым А. М. Бутлеровым, помимо таких, уже известных классов соединений, как н-алканы, первичные и вторичные спирты, должны были существовать также изоалканы и третичные спирты. Успешный синтез соединений этого типа явился одним из важнейших свидетельств справедливости основных концепций структурной теории.
Для проверки теории ароматичности в свое время было исключительно важным ответить на вопрос о том, является ли ароматическим соединением циклооктатетраен (71, схема 1.22), ближайший аналог бензола? Синтез этого ранее неизвестного вещества, выполненный Вильштеттером в 1911 г., и изучение его свойств позволили дать однозначный ответ на этот вопрос (циклооктатетраен — неароматичен).
Схема 1.22
He менее важным для подтверждения представлений структурной теории явился синтез таких соединений, как оптически активные аллены 72 или четвертичные аммониевые соли 73, т.е. типов структур, для которых возможность оптической изомерии непосредственным образом следовала из представлений Вант-Гоффа и Ле Беля.
Для химика-органика такие структуры, как кубан (74), призман (75) или суперфан (76), исполнены столь неотразимого очарования, что просто невозможно было не попытаться их синтезировать. Успешное осуществление синтезов этих и других соединений с не менее экзотическими структурными каркасами не только открыло совершенно новые области исследования для экспериментаторов, но и стимулировало разработку новых представлений теоретической органической химии.
Синтез ротаксанов (77), в которых циклическая молекула “надета” на линейную и не может с нее “соскользнуть” из-за наличия объемистых концевых групп, и катенанов (78), в которых две циклические молекулы связаны друг с другом, как звенья в цепи, разрешил положительно вопрос о возможности существования молекул, фрагменты которых соединены без помощи ковалентных связей (схема 1.23).
Схема 1.23
С развитием органической химии проблемы дизайна и синтеза соединений новых структурных типов, отнюдь не иссякают. На схеме 1.23 приведены также формулы тетраэдрана (79) и фенестрана (80) как примеры структур, все еще остающихся проблемными для синтеза.
Более подробно ряд общих и конкретных аспектов молекулярного дизайна, направленного на создание необычных и теоретически интересных структур, рассмотрен в гл. 4 данной книги.
1.5.3. Расширение круга известных органических соединений
Это — одна из традиционных и наиболее скромных сторон деятельности химиков-синтетиков. Скромных потому, что большинство таких синтезов носит весьма заурядный характер, и уже давно никого не удивляют работы, итогом которых является синтез десятков или даже сотен новых веществ.Понятно, что такой путь саморазвития был естественным и необходимым в эпоху становления органической химии. Однако в настоящее время, когда описаны миллионы органических соединений и их основные классы изучены достаточно подробно, такой “синтез ради синтеза” может показаться недопустимым излишеством. Стоит ли, в самом деле, отвлекать силы от целенаправленных исследований на синтез еще миллионов* новых соединений, незная, зачем они могут понадобится, вернее, даже зная наверняка, что сведения о большинстве из них просто застынут без движения в справочниках?
* К настоящему времени уже получено около 15 млн. органических соединений. Темп расширения этого “ассортимента” (примерно 500 новых соединений в день) пока не обнаруживает тенденции к снижению.Как и все фундаментальные науки, органическая химия исследует неизвестное. Поэтому не представляется возможным предсказать открытия в той или иной ее области (или их невозможность) и тем более практическую значимость будущих открытий. С уверенностью, однако, можно утверждать, что если остановить развитие органической химии вширь, то не будет открытий как новых областей исследования, так и новых возможностей применения получаемых веществ.Химики, получившие более 100 лет назад бензоат холестерина — типично рутинный (даже для того времени) синтез нового производного хорошо известного соединения, не могли подозревать, что открывают путь к созданию невероятного разнообразия устройств, в которых применяются жидкие кристаллы — новое состояние вещества, которое неожиданно было открыто на примере бензоата холестерина. Вспомним также, что составившее эпоху в химиотерапии открытие сульфаниламидных препаратов явилось абсолютно непредсказуемым следствием широких исследований, направленных на синтез сотен и сотен ароматических производных, потенциально полезных для создания новых азокрасителей.
Типичным примером искусственного создания совершенно новой области для исследования может служить химия фторорганических соединений.
Эта область возникла из чисто академического вопроса, сродни детскому любопытству: а как будут выглядеть органические соединения, если в них все большее число атомов водорода замещать на атомы фтора? В свое время (в 1920—30-х годах) это была довольно трудоемкая область исследования, и сложность синтеза перфторированных органических соединений, казалось бы, навсегда предопределяла их судьбу — остаться в сфере интересов “чистой науки”, без перспектив практического использования. Однако именно в этой области исследователей ожидали не только открытия в области теории, но и появление новых классов веществ с уникальными физико-химическими свойствами.
Среди этих веществ следует упомянуть фторопласты [34], полимеры с исключительным набором полезных свойств, не заменимые в этом отношении никакими из известных природных или искусственных материалов; фреоны, на протяжении десятилетий служившие основой холодильной и аэрозольной техники; перфторированные производные типа перфтортетрагидрофурана, неожиданно оказавшиеся великолепными растворителями — переносчиками кислорода (на основе последних и были разработаны искусственные кровезаменители, знаменитая “голубая кровь”).
Несколько позднее была открыта еще одна область возможного практического применения фторпроизводных, на этот раз в медицине. Было обнаружено, что фторсодержащие аналоги природных метаболитов, которые почти неотличимы от нефторированных соединений по своим базовым структурным характеристикам, являются хорошими антиметаболитами — ингибиторами соответствующих ферментных систем, так что результатом их воздействия на клетку является блокирование определенных биохимических функций. Многие сотни такого рода соединений были синтезированы и использованы в биохимических и медицинских исследованиях [35]. Один из наиболее известных представителей этого семейства, 5-фторурацил (фторированный аналог одного из нуклеиновых оснований, остатки которых входят в состав ДНК), нашел применение в качестве высокоактивного противоопухолевого препарата.
Искусственно созданные органические вещества могут служить также источником открытий в областях науки, казалось бы, никак не связанных с органической химией.
Наглядным примером могут служить работы, направленные на создание органических проводников и сверхпроводников. Неспособность типичных органических соединений проводить электрический ток известна с давних пор. Действительно, именно изолирующие свойства полимеров обусловили их широчайшее внедрение в практику в качестве всевозможных покрытий. Однако в последние десятилетия было найдено, что некоторые типы полимеров могут проявлять свойства проводников. Так, полимеры общей формулы -(СН=СН)n-, получаемые полимеризацией ацетилена в условиях реакции Циглера—Натта, приобретают свойства металлических проводников при допировании (частичном окислении мягкими окислителями типа иода). Электропроводность допированного полиацетилена может быть очень значительной (104 См/см), всего лишь на два порядка меньше, чем, например, у серебра(106 См/см; ср. с величиной 10-18 См/см для почти идеальнoro изолятора, тефлона). Важность этого открытия была очевидной, и за ним последовал взрывоподобный рост активности в области поиска других органических соединений с подобными свойствами [36] *. Помимо полиацетиленов, другие полимеры, содержащие длинные сопряженные цепи, такие, как полифенилен, полипиррол или полианилин**, также обнаружили способность проходить электрический ток в различных условиях [37].
* Дальнейшее развитие работ в области допированных полиацетиленов оказалось чрезвычайно успешным, свидетельством чего может служить присуждение Нобелевской премии по химии за 2000 г. А. Хигеру, А. Мак-Диармиду и X. Сиракаве за “Открытие и развитие области электропроводящих полимеров” (подробнее об этом можно прочесть в статье: Кобрянский В. М., Природа, № 1, с. 7, 2001).** Уместно заметить, что полианилин был получен еще в 1862 г., но до недавнего времени никто и не подозревал о его довольно высокой электропроводности (200 См/см).В исследованиях несколько иного плана было обнаружено еще более интересное физическое свойство органических соединений, а именно способность некоторых органических веществ служить сверхпроводниками. Так, например, комплексы с переносом заряда тетратиафульвалена (81) и тетрацианохинодиметана (82) состава 1:1 (схема 1.24) способны не только проявлять свойства металлических проводников при комнатной температуре, но и становятся сперхпроводниками при низких температурах. Синтезированы и изучены многочисленные соединения этого и сходных типов. Среди них особенно интересными оказались комплексы с переносом заряда, полученные из бис(этилендитио)тетратиафульвалена и неорганических анионов. Некоторые из этих комплексов обнаруживали сверхпроводящие свойства при температуре 10, 4 К.
Схема 1.24
Хотя рассмотренные выше результаты еще не позволяют говорить о применении органических металлов как о немедленной практической перспективе, они, тем не менее, позволяют вести в дальнейшем уже не случайный, а целенаправленный поиск соединений, обладающих требуемыми структурными характеристиками. Таким образом, в число целей органического синтеза оказывается включенной задача получения структур, оптимальным образом приспособленных для решения чисто физических проблем — задача, которая еще недавно находилась исключительно в поле компетенции неорганической химии и собственно физики.
В заключение хочется сделать еще одно замечание, касающееся своеобразия органического синтеза. Присущий этой области науки созидательный характер проявляется еще и в том, что здесь любой грамотный результат, в том числе и неудачный с точки зрения первоначального замысла, представляет собой вклад в сокровищницу человеческих знаний. Действительно, синтез нового соединения, независимо от того, отвечают или нет его свойства ожиданиям экспериментатора, в любом случае остается синтезом нового, ранее неизвестного объекта природы, т. е. открытием *.
* Сказанное вовсе не означает, что авторы настаивают на научной значимости любой работы, результатом которой является синтез нового вещества. Именно отмеченная выше бесконечность многообразия возможных структур органических соединений заставляет особенно требовательно относится к выбору области синтетического поиска, и исследование в этой области может считаться оправданным только при условии четкой формулировки его цели.В настоящей главе рассмотрены лишь некоторые из общих вопросов, относящихся к целям и задачам органического синтеза. Читателю, желающему более глубоко познакомиться с общими вопросами истории, философии и методологии, а также мотивации органического синтеза, мы можем порекомендовать книгу Хоффмана «Такой одинаковый и разный мир» [38]. Чрезвычайно интересный материал, относящийся как к достижениям современного органического синтеза, так и к рассмотрению основных тенденций его развития имеется в великолепно написанном обзоре Зибаха под названием «Органический синтез. Что дальше?» [39].
ЛИТЕРАТУРА
1. (а) Более подробно о химической истории “королевского” пурпура см. обзор: McGovern P.Е., Michel R.Н. Асc. of Chem. Res., 23, 152 (1990); см. также:Hoffmann R. American Scientist, 78, 308 (1990);
(b)Bayer A.Ber., 11, 2128 (1878).2. Grebe С., Lieberman С. Ber., 2, 332 (1869).
3. Корана X.Г., цитировано по лекции: “Полный синтез биологически функционального гена”, в сб.: “Итоги и перспективы развития биоорганической химии и молекулярной биологии”, Овчинников Ю.А., Колосов М.Н. (ред.). — М.: Наука, 1978, cc. 203 — 209.
4. Reichstein Т.A., Gussner A. Helv.Chim. Acta, 18, 608 (1935).
5. (a) Bergstrom S. Science, 157, 382 (1967);
(b) Обзоры в сб.:Curtis-Prior P.В.(ed.), Prostaglandins: Biology and Chemistry of Prostaglandins and Related Eicosanoids, Churchill Livingstone, Edinborough, 1988.6. (а) Популярное изложение истории таксола см.: Bowman S.Chem.Eng. News, 1991, Sept. 2, 11; см. также: Freemantle М., там же, 1994, Aug. 1;
(b) обзоры см.: Guenard D., Gueritte-Voegelein F., Potier P. Acc. Chem. Res., 26, 160 (1993); Nicolaou К.С., Guy R.К., Dai W.M. Angew. Chem., Int. Ed. Engl., 33, 15 (1994);
(с) Holton R.A., Kim Н. B., Somow C., Liang F., Biediger R.J., Boatman P.D. Shindo M., Smith C.C., Kirn S., Nadizadeh H., Suzuki Y., Too C., Vu, P., Tang S., Zhang P., Murthi K.K., Gentile L.N., Liu J. H. J. Am. Chem. Soc., 116, 1599 (1994);
(d) Nicolaou K.C., Claiborne C.F., Nantermet P.G., Couladorous Е.A., Sorensen Е.J. J. Am. Chem. Soc., 116, 1599 (1994);
(е) см., например:Blechert S., Kleine-Klausing A. Angew. Chem., Int. Ed. Engl., 30, 412(1991).7. (а) О проблеме иммуномодуляторов см.:Kessler H., Mierke D.F., Donald D., Furber M. Angew. Chem., Int. Ed. Engl., 30, 954 (1991);
(b) об истории проблемы см.:Stinson S. Chem. Eng. News, 1989, Feb. 6, 30;
(с) Jones Т.К., Mills S.G., Reamer R.A., Askin D., Desmond R., Ryan K.M., Volante R.P., Shinkai I.J. Am. Chem. Soc., 111, 1157 (1989);
(d) Nakatsuka M., Ragan J. A., Sammakia Т., Smith D.B., Uehling D.E., Schreiber S.L., J. Am. Chem. Soc., 112, 5583 (1990);
(е) Goulet М.Т., Hodkey D.W. Tetrahedron Lett., 32, 4627 (1991);
(f) Andrus M.B., Schreiber S.L. J. Am. Chem. Soc., 115, 10420 (1993).8. (а) Обзор см.:Siddal J.В. Chemical Aspects of Hormonal Interactions, Chemical Ecology, в сб.: Chemical Ecology, Academic Press Inc., New York, 1970, Ch. 11, p. 281;
(b) Trost B.M. Acc. Chem. Res., 3, 120 (1970).9. (a) Gibberellins and Plant Growth, Krishnamurthy H. N. (ed.), Wiley, New York, 1975;
(b) Corey E.J., Danheiser R.L., Chandrasekaran S., Keck G.E., Gopalan B., Larsen S.D., Siret, P., Gras J.L. J. Am. Chem. Soc., 100, 8034 (1978).10. Addicott E.Т., Lyon J.L., Ohkuma K., Thiessen W.E., Cams H.R., Smith O.E., Cornforth J.W., Millborrow B.V., Ryback G., Wareing P.F. Science, 159, 1493 (1968).
11. (a) Butenandt A., Hecker E., Hopp M., Koch W. Ann., 658, 39 (1962);
(b) Truscheit E., Eiter K. Ann., 658, 65 (1962).12. Обзор см.:Kelly D.R., Chemistry in Britain, 1990, 124.
13. (а) Выделение см.: Butler С.G., Callow R.K., Johnston N.C. Nature, 184, 1871 (1959);
(b) синтез см.:Bellassoued M., Majidi A. Tetrahedron Lett., 32, 7253 (1991) и цитированные в этой статье работы.14. Обзор см.:Pheromones, Birch М.С. (ed.), North-Holland Research Monographs, Frontiers in Biology, v.32, 1977, 595 pp.; см. также научно-популярные статьи: “Slave-making ants”, Topoff H. American scientist, 78, 520 (1990); “Empire of the ants”, Wilson E. D. Discoverer, № 3, 44 (1990).
15. (а) Об истории открытия и механизме действия, см.:Jaenicke L., Boland W. Angew. Chem., Int. Ed. Engl., 21, 643 (1982);
(b) синтез: Abraham W.D., Cohen T. J. Am.Chem.Soc., 113, 2313 (1991); Grouse G.D., Paquette L.A. J.Org.Chem., 46, 4272 (1981) и цитированные в этой статье работы.16. Синтез см.:Corey Е.J., Achiwa К., Katzenellenbogen J. A. J. Am. Chem. Soc., 91, 4318 (1969).
17. MacMorris Т.С., Barksdale A.W. Nature, 215, 320 (1967); Arsenault G.P., Biemann K., Barksdale A.W., MacMorris T.C. J. Am. Chem. Soc., 90, 5635 (1968).
18. (а) Выделение см.: Cook С. Е., Whichard L.P., Turner В., Wall М.Е., Egley Е.Н. Science, 154, 1189 (1966);
(b) синтез см.:Johnson A.W., Govda G., Hassanali A., Knox J., Monako S., Razavi Z., Rosebery G. J. Chem. Soc., PerkinTrans. 1, 1981, 1734;
(с) Chang M., Netzly D.H., Butler L.G., Lynn D.G. J. Am. Chem. Soc., 108, 7858 (1986).19. Подробнее об этом см.:Барбье М. Введение в химическую экологию. — М.: Мир, 1978, 229 с.; см. также: Levinson G. Naturwissenshaften, 59, 477 (1972); Gerout V., в сб.: Progress in Phytochemistry, v.2, J. Wiley& Sons, London, 1970, p. 143.
20. Обзор по антифидантам и возможностям их использования в защите растений см.: Ley S.V., Toogood P.L. Chemistry in Britain, 1990, January, 31.
21. Eisner Т., Meinwald J. в сб.:Pheromone Biochemistry, Prestwich G.D., Bloomquist G.J. (eds.), Academic Press, Orlando, 1987, ch. 8, p. 251; очень живое изложение истории этих работ можно найти в популярной статье:Meinwald J. Engineering & Science, 49, № 5, 14 (1986).
22. (a) Corey E.J., Dittami J.P. J. Am.Chem.Soc., 107, 256(1985);
(b) Corey E.J., Gиzтап-Perez A., Noe M.C. J. Am. Chem. Soc., 116, 12109 (1994).23. Об истории открытия см.: Roberts R.М. Serendipity, J. Wiley, New York, 1989, ch. 24, p. 159.
24. (a) Sarrett, L.H. J. Biol. Chem., 162, 591(1946);
(b) краткая история работ по созданию стероидных препаратов изложена в обзоре:Hirschman R. Angew. Chem., Int. Ed. Engl., 30, 1278 (1991);
(с) обзор см.:Шоппи К.У. “Стероиды”, в сб.: Перспективы развития органической химии. — М.: ИЛ, 1959, с. 223.25. (а) Обзор см.:Уокер Д. “Химиотерапия”, ссылка [24с], с. 301;
(b) Klayman D.L. Science, 228, 1049 (1985);
(с) см., например: Avery М.A., Chong W.К.М., Jennings-White С. J. Am. Chem. Soc., 114, 974 (1992); см. также: Ye B., WuY.-L. J. Chem. Soc., Chem. Comm., 1990, 726;
(d) Posner G.H., Oh C.H., Milhous W.K. Tetrahedron Lett., 32, 4235 (1991) и цитированные в этой статье работы.26. (a) Still W С. J. Am. Chem. Soc., 101, 2493 (1979);
(b) Hauptman H., Muhlbauer G., Sass H. Tetrahedron Lett., 27, 6189 (1986);
(с) Mori М., Okada К., Shimazaki К., Chuman Т. Tetrahedron Lett., 31, 4037 (1990).27. (а) Популярное изложение истории открытия и исследования см.:Stinson S. Chem. Eng. News, 1995, Apr. 17, 22;
(b) Barrett A.G.M., Kasdorf K., Tustin G.J., Williams D.J. J. Chem. Soc., Chem. Comm., 1995, 1143;
(с) Kuo M.S., Zielinsky R.J., Cialdella J.I., Marschke C.K., Dupuis M.J., Li G.P., Kloosterman D.A., Spilman C.H., Marshall V.P. J. Am. Chem. Soc., 117, 10629 (1995).28. (а) Основные ссылки и общий обзор см.: Cross P.Е., Dickinson R.P. Chemistry in Britain, 1991, 911;
(b) Bhagwat S.S., Hamann P.R., Still W.C., Buntings, Fitzpatrick F.A. Nature, 315, 511 (1985);
(с) Bundy G.L., Tetrahedron Lett., 1975, 1957; см. также: Nicolaou К.С., Magolda R.L, Smith J.B., Aharony D., Smith E.F., Lefer A.M. Proc. Natl. Acad.Sci. USA, 76, 2566 (1979).29. Вудворд P.Б. “Синтез”, ссылка [24с], C. 119.
30. Corey E.J., Angew. Chem., Int. Ed. Engl., 30, 455 (1991).
31.(a) Gomberg M., J. Am. Chem. Soc., 22, 757 (1900);
(b) обзор см.:Gomberg M. Chem. Rev., 1, 91 (1925);
(с) Lankamp H., Nauta W.Т., MacLean C. Tetrahedron Lett., 1968, 249:
(d) изложение поучительной истории загадки гексафенилэтана можно найти в обзоре: McBride J.М. Tetrahedron, 30, 2009 (1974).32. (a) Sachse H. Ber., 23, 1363 (1890);
(b) Barton D.H.R. Experientia, 6, 315 (1950);
(с) Бартон Д.P. «Стереохимия», ссылка [24с], с. 57.33. Цитировано по ссылке [29], с. 133.
34. История открытия тефлона увлекательно изложена в книге: Roberts R.М. Serendipity. Accidental Discovery in Science, J. Wiley & Sons, New York, 1989, p. 187.
35. Характерные примеры можно найти в обзорах: Mann J. Chem. Soc. Rev., 16, 381 (1987); Welch J.Т. Tetrahedron, 43, 3123 (1987).
36. Краткое изложение истории основных направлений в изучении проводящих полимеров см.: Kanatudis М.G. Chem. Eng. News, 68, № 49, 36 (1990).
37. Обзор см.: Williams J.M., Веnо M.A., Wang H.H., Leung P.C.W., Emge T.J., Geiser U., Carlson K.D. Acc. Chem. Res., 18, 261 (1985).
38. Hoffmann R. The Same and Not the Same, Columbia University Press, New York, 1995 (имеется перевод: Хоффман P. Такой одинаковый и разный мир. — М.: Мир, 2001).
39. Seebach D. Angew. Chem., Int. Ed. Engl., 29, 1320 (1990).
 |
Публикуется с любезного разрешения авторов и издательства "Мир" |
![]()
Январь 2002