|
ВЕСТНИК РОССИЙСКОЙ АКАДЕМИИ
НАУК
том 73, № 11, с. 987-994 (2003) |
|
ВЕСТНИК РОССИЙСКОЙ АКАДЕМИИ
НАУК
том 73, № 11, с. 987-994 (2003) |
© М.С. Гельфанд, В.А. Любецкий
БИОИНФОРМАТИКА:
ОТ ЭКСПЕРИМЕНТА К КОМПЬЮТЕРНОМУ АНАЛИЗУ
И СНОВА К ЭКСПЕРИМЕНТУМ. С. Гельфанд, В. А. Любецкий
Примерно в середине 50-летия, отделяющего нас от открытия структуры двойной спирали ДНК, в молекулярной биологии произошел мощный технологический прорыв: Ф. Сэнгер, Ф. Максам и В. Гильберт предложили методики быстрого секвенирования ДНК, то есть установления последовательности нуклеотидов в геноме. Уже в 1978 г. было опубликовано 200 статей, описывавших секвенированные нуклеотидные последовательности, затем объем этих данных стал расти в геометрической пропорции (рис. 1). Были сделаны наблюдения, изменившие устоявшиеся представления о линейной последовательности генов в ДНК: перекрывающиеся гены, сплайсинг и альтернативный сплайсинг (механизм порождения множественных РНК, соответствующих одному и тому же гену), рекомбинация генов иммуноглобулинов.
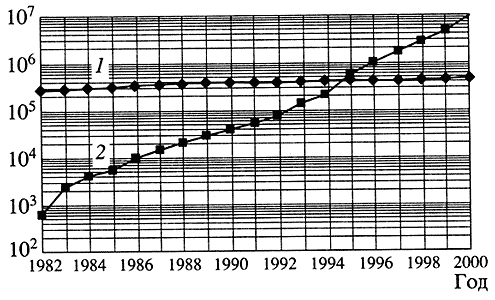
Рис. 1. Количество статей в библиографической базе данных PubMed (1)
и количество фрагментов в банке данных нуклеотидных последовательностей GenBank (2) в 1982-2000 гг.
Данные приведены в логарифмическом масштабеСущественную роль в развитии геномных подходов сыграли банки нуклеотидных последовательностей. Довольно быстро стало понятно, что невозможно сопоставлять последовательности, сравнивая вручную длинные ряды букв, приводимых на рисунках к статьям. Уже в 1979 г. было начато обсуждение того, как хранить последовательности ДНК и РНК и как обеспечивать доступ к ним. Первые выпуски банков данных GenBank (США) и EMBL (Европа) появились в 1982 г., и уже в следующем году они сыграли существенную роль в биологической работе: сходство последовательностей онкогена v-sis из вируса саркомы обезьян и фактора роста тромбоцитов, обнаруженное при сравнении новосеквенированного гена со всеми опубликованными, послужило основой для гипотезы о сходстве воздействия онкогенов и нормальных клеточных белков, экспрессирующихся на определенных стадиях жизни клетки [1]. С тех пор сравнение новой последовательности с последовательностями из банка данных стало рутинным элементом работы с любым геном, а помещение каждой новой последовательности в банк - необходимым условием журнальной публикации.
Анализ нуклеотидных последовательностей привел к революционным изменениям во многих областях биологии, в частности, в таксономичес-ких представлениях о механизмах эволюции геномов. Например, оказалось, что геномы прокариот крайне нестабильны: многие гены в эубактериях и архебактериях подвержены горизонтальному переносу, причем это свойственно не только генам, кодирующим ферменты и транспортеры (что можно было бы объяснить приспособлением бактерий к различным экологическим нишам), но и генам, кодирующим такие фундаментальные белки, как аминоацил-тРНК синтетазы, белки рибосом и ферменты системы репарации [2]. В связи с этим ряд исследователей предлагает вообще отказаться от понятия эволюционного дерева применительно к прокариотам, рассматривая взамен что-то вроде направленной сети. Тем самым степень родства двух организмов будет определяться не только временем их расхождения от общего предка, но и долей генома, которую они сохранили от него.
Дальнейший прогресс технологии, в частности, разработка К. Муллисом в 1986 г. полимеразной цепной реакции и создание тогда же первых удовлетворительно работающих приборов автоматического секвенирования Л. Худом и Т. Хункапиллером, привел к тому, что сразу в нескольких странах началось обсуждение возможности полного секвенирования генома человека. В 1988 г. соответствующий проект стартовал в США и практически тогда же - в СССР. Уже при разработке проекта были приняты критически важные решения, оказавшие существенное влияние на дальнейшее развитие геномики. Первое из них состояло в том, чтобы секвенировать не только геном человека, но и геномы модельных организмов: нематоды Caenorhabditis elegans, плодовой мухи Drosophila melanogaster, дрожжей Saccharomyces cerevisiae, растения Arabidopsis thaliana, бактерий Escherichia coli. Bacillus subtilis и других. При выборе объектов секвенирования в основном учитывался баланс между изученностью организма и размером его генома. В результате стал возможен сравнительный анализ сразу многих геномных данных.
Второе столь же важное решение состояло в том, что данные секвенирования геномов немедленно становились доступными мировому научному сообществу. В 1996 г. были сформулированы "Бермудские принципы" (названы по месту проведения конференции), согласно которым даже небольшие фрагменты геномов, полученные в рамках проекта "Геном человека" и аналогичных программ, сразу же помещались в банки данных и могли быть использованы всеми желающими. Одновременно в журналах публиковались результаты анализа больших секвенированных фрагментов геномов и целых хромосом. Такая ситуация иногда приводила к недоразумениям: группы, занимающиеся секвенированием, опасались, что кто-то другой опубликует существенные результаты на основе анализа полученных ими данных. Но когда геномные проекты начали реализовываться частными компаниями, ряд ведущих журналов отказался от требования помещать последовательности в стандартные банки данных.
В середине 80-х годов прошлого века начало складываться новое научное направление, названное биоинформатикой, или вычислительной молекулярной биологией. В рамках этого направления развиваются алгоритмы для анализа последовательностей биополимеров (ДНК и белков) и их пространственной структуры, строятся модели метаболизма и регуляторных взаимодействий. Затем они применяются для решения биологических задач.
Важность развития биоинформатики диктуется несколькими обстоятельствами. Самое простое из них - это объем геномной информации (рис. 2), делающий невозможным ее ручную обработку без использования алгоритмических методов. Современные технологии геномики и протеомики немыслимы без интенсивного применения компьютерной обработки результатов. Но дело не только в этом. Во многих случаях сопоставление геномных данных позволяет делать новые и совершенно нетривиальные выводы, которые затем могут быть проверены экспериментально. За последние несколько лет биоинформатика стала самостоятельной областью на стыке биологии и математики со своими специфическими задачами и методами их решения [3]. Она использует методы математической логики и теории алгоритмов, информационных и стохастических процессов, теории динамических игр и статистики и т.д.
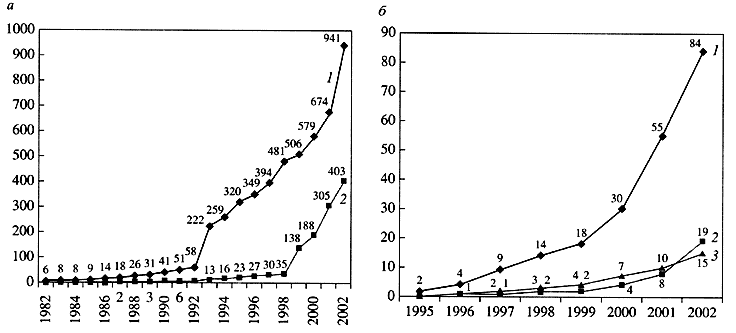
Рис. 2. Количество полных геномов в банке нуклеотидных последовательностей GenBank [а - геномы вирусов (1) и органелл (2); б - геномы бактерий (1), эукариот (2) и архебактерий (3)]; резкие всплески в 1993 г. (вирусы) и в 1999 г. (органеллы) связаны не с увеличением числа опубликованных геномов, а с изменением структуры банкаСамым простым компьютерным методом считается сравнение вновь полученной последовательности с уже имеющимися в банках данных. Часто оказывается, что можно найти похожий уже изученный белок, благодаря чему удается предсказать функцию нового белка. Если близкие гомологи отсутствуют, применяют более тонкие методы анализа. В частности, существуют алгоритмы поиска в белковых последовательностях трансмембранных сегментов и сигнальных пептидов, основанные на анализе статистических особенностей этих структурных элементов. Используя такие алгоритмы, можно предсказать, локализуется ли белок в цитоплазме, в мембране или секретируется из клетки. Анализ больших групп белков, имеющих одну и ту же функцию (например, АТФазную активность), позволил создать библиотеки функциональных подписей, то есть коротких аминокислотных последовательностей, разделенных вариабельными промежутками и соответствующих функциональному сайту в пространственной структуре белка. Такие подписи могут быть общими даже для белков, не демонстрирующих видимого сходства на уровне всей последовательности, и их обнаружение в исследуемом белке может указать .на биохимическую функцию последнего, даже если для него не найдется экспериментально изученных гомологов.Наибольшую силу компьютерные методы показали при анализе полных геномов: сначала бактерий (гемоглобинофильная палочка Haemophilus influenzae в 1995 г.), а вскоре - архебактерий и эукариот (дрожжи Saccharomyces cerevisiae в 1996 г.). В настоящее время доступно более ста полных геномов самых разнообразных организмов, в основном бактерий (см. рис. 2, б), и ясно, что подавляющее большинство из них не может быть подробно исследовано в экспериментальной лаборатории. Однако оказалось, что в общих чертах удается описать физиологию организма путем чисто компьютерного анализа его генома [4].
Такой анализ начинается с картирования генов. При этом используются статистические методы, опирающиеся на различия в свойствах белок-кодирующих и некодирующих областей, анализ сигналов на границах генов, а также сравнение с уже изученными генами. Можно считать, что задача картирования генов в геномах прокариот практически решена, в то время как точность таких предсказаний в геномах эукариот еще недостаточная, хотя есть надежда, что она будет расти по мере увеличения количества доступных для анализа геномов, находящихся в разной степени родства. К тому же появились новые методы сравнения геномных последовательностей, которые базируются на том, что белок-кодирующие гены меняются в ходе эволюции медленнее, чем окружающие их некодирующие области. При сравнении геномных последовательностей эти гены видны как островки сходства на фоне сильно изменившихся некодирующих областей. Существенно, что такой анализ (и его более простой вариант, применяемый при работе с бактериальными геномами) дает возможность обнаруживать совершенно новые гены, белковые продукты которых не имеют известных гомологов.
Вслед за этим проводится функциональная аннотация белков. Оказывается, что таким образом удается детально охарактеризовать от половины до двух третей бактериального генома, функции еще 10-15% генов устанавливаются в общих чертах. Для эукариот подобные оценки труднее дать как в силу уже упомянутых проблем с картированием их генов, так и из-за наличия большого количества дуплицированных генов с одинаковой биохимической функцией, но с различной ролью в жизни клетки (например, факторов транскрипции или протеин-киназ, участвующих в различных регуляторных каскадах). Все же сделанные предсказания оказываются достаточными для предварительной метаболической реконструкции. С этой целью устанавливается соответствие между предсказанными белками и универсальной картой метаболических путей, суммирующей данные о всех химических реакциях, когда-либо наблюденных в живой клетке любого организма. Такая проекция генома на карту метаболических путей позволяет описать основные физиологические характеристики организма.
Предварительная метаболическая реконструкция, основанная лишь на анализе белковых гомологий, как правило, содержит белые пятна: нелогичные разрывы метаболических путей, тупики и т.п. Более того, заметная доля реакций на универсальной метаболической карте не соответствует никаким известным белкам. В то же время примерно 10% типичного генома - это высоко-консервативные гены с неизвестной функцией, присутствующие во многих геномах. Отметим, что само существование таких белковых семейств было открыто только после анализа нескольких полных геномов. Открытие оказалось достаточно неожиданным: до того господствовала точка зрения, согласно которой основные (и потому присутствующие во многих геномах) белки уже известны, а новые белки будут, как правило, геном-специфичными. Естественно, что сразу же многие группы исследователей начали работу по выяснению функций этих консервативных белковых семейств.
Итак, после предварительной метаболической реконструкции можно идентифицировать пробелы как универсальные, так и специфичные для данного генома, после чего встает задача заполнения этих пробелов. Поскольку проводится анализ полного генома, нет оснований ожидать, что недостающие гены еще будут найдены.
Для заполнения пробелов применяются различные методы геномного анализа, разработанные в последние годы, в том числе позиционный. Было замечено, что гены, располагающиеся на хромосоме рядом, часто кодируют функционально связанные белки, например, ферменты, катализирующие последовательные стадии какого-то метаболического пути, либо транспортный белок, отвечающий за импорт какого-либо вещества, и ферменты, это вещество перерабатывающие. Разумеется, близость генов в одном геноме или в группе очень близких геномов мало что значит. Но если какие-то гены близки (кластеризуются) в хромосомах нескольких достаточно далеких геномов, то это уже можно рассматривать как важное свидетельство в пользу наличия функциональных связей между продуктами этих генов.
Несмотря на внешнюю банальность позиционного подхода, он оказался весьма плодотворным и позволил закрыть заметное число пробелов как в отдельных геномных метаболических реконструкциях, так и на универсальной карте метаболических путей. Например, с помощью позиционного анализа был идентифицирован ген архебактериальной шикимат-киназы. Более того, оказалось, что используя позиционный анализ, можно закрыть пробелы и в метаболических путях эукариот. Для этого сначала методом анализа белковых гомологий выделяют соответствующий бактериальный метаболический путь, идентифицируют в нем пропущенный ген, а затем определяют его гомолог в исследуемом эукариотическом геноме. Так, в частности, была выделена НМН/НаМН-аденилилтрансфераза человека и изучена ее пространственная структура, что важно для понимания механизма действия антиракового препарата тиазофурина [5].
В одном из вариантов позиционного анализа, особенно полезном для изучения метаболических путей и белок-белковых взаимодействий эукариот, рассматриваются случаи слияния генов. Если в каком-то геноме два гена слились и кодируют одну полипептидную цепь, - это веское свидетельство в пользу того, что кодируемые генами белки всюду находятся в физическом или функциональном взаимодействии. Однако при таком анализе следует относиться с осторожностью к разделению ортологов (соответствующих генов в разных геномах) и паралогов (дуплицированных генов).
Другой важный метод вычислительной гено-мики основан на анализе сигналов, регулирующих экспрессию генов. Опять-таки, наборы совместно регулируемых генов, как правило, образуют функционально связанные группы. Такой группой может быть метаболический путь, включающийся при недостатке какого-то вещества, система ответа на внешний раздражитель (скажем, тепловой шок) или система контроля физиологического состояния клетки (например, переход к споруляции при голодании у бацилл). Если удается выделить сигнал в последовательности ДНК, отвечающий за регуляцию подобной группы генов, то можно построить распознающее правило и искать другие гены, имеющие тот же сигнал и, стало быть, регулируемые совместно с рассматриваемой группой. Анализ регуляции важен и сам по себе, поскольку он позволяет ответить не только на вопрос: "Что клетка может делать?", но и на вопрос: "В каких условиях она это делает?".
И все же в большинстве случаев не удается построить удовлетворительно работающее правило для распознавания регуляторных сигналов. Тогда полезным оказывается одновременный анализ многих геномов. Дело в том, что наборы совместно регулируемых генов, соответствующие функциональным подсистемам, консервативны. В результате истинные регуляторные сигналы обнаруживаются перед гомологичными генами сразу во многих геномах, в то время как ложные сигналы располагаются случайным образом. Это позволяет фильтровать предсказанные сигналы на основе требования согласованности предсказаний, тем самым повышая надежность каждого отдельного сигнала.
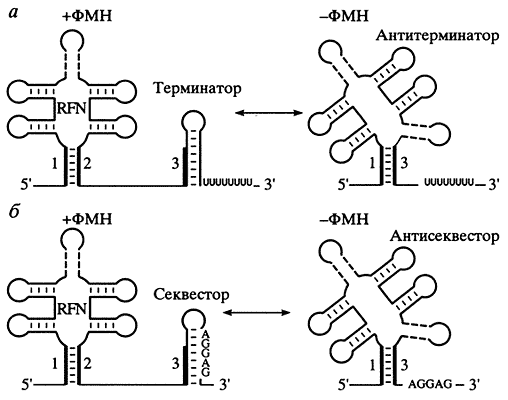
Такой подход дает возможность не только искать новые регулируемые гены в изучаемых функциональных системах, но и описывать совершенно новые регуляторные механизмы. Одним из них стала исследованная нами система регуляции биосинтеза витаминов у бактерий (рис. 3).
Рис. 3. Альтернативные вторичные структуры в 5'-нетранслируемых областях матричной РНК, регулирующие работу генов биосинтеза рибофлавина в зависимости от концентрации флавинмононуклеотида (ФМН) [6]Сравнительный анализ большого числа геномов позволил выделить и описать регуляторные сигналы, консервативные даже при очень далеких сравнениях (вплоть до архебактерий) [6]. Хотя сами сигналы в разных системах (рибофлавин, тиамин, кобаламин) различны, они обладают рядом общих свойств. Например, в отличие от известных аттенюаторов *, сигналы такого типа действуют на разные процессы: терминацию транскрипции (преимущественно у грамположитель-ных бактерий) и инициацию трансляции (у грам-отрицательных бактерий). Эта модель, получившая название РНК-переключателя, затем подтвердилась в эксперименте [7]. Более того, оказалось, что образование альтернативных структур регулируется непосредственно связыванием витамина - концевого продукта метаболического пути. Аптамеры (структуры РНК, связывающие малые молекулы) были известны и ранее, но они наблюдались in vitro, исследованные же системы стали первыми примерами природных аптамеров. Сигналы такого типа найдены и у эукариот, так что, возможно, РНК-переключатель - древнейшая известная система регуляции экспрессии генов. Массовый поиск аттенюаторных регуляторных сигналов был продолжен [8]. Еще раз подчеркнем: сравнительный подход позволяет не просто описывать новые примеры уже известных объектов - функций белков, регуляторных сигналов, метаболических путей, - но и обнаруживать совершенно новые явления.При наличии ФМН образуются регуляторные шпильки - RFN и терминатор, препятствующие экспрессии оперона, при его отсутствии - альтернативная конформация - антитерминатор; 1,2,3- участки матричной РНК, способные к формированию альтернативной вторичной структуры; а -аттенюация транскрипции за счет формирования терминатора; 6 - аттенюация трансляции за счет ингибирования ее инициации (так как участок Шайно-Дальгарно AGGAG находится в спаренном состоянии)
* Регуляторная система, основанная на формировании альтернативных вторичных структур РНК.Итак, сравнительный компьютерный анализ становится мощным средством в руках биолога. Существенно, что в арсенале биоинформатики много разнообразных методов, которые позволяют многократно проверять полученные результаты на их согласованность и непротиворечивость. Таким образом, факты, выявленные в ходе компьютерного анализа, могут считаться столь же надежными, как и факты, установленные в эксперименте, - разумеется, при наличии достаточных проверок и хорошем контроле. Резкая грань, проходившая ранее между предварительным теоретическим анализом и последующей экспериментальной проверкой, стирается, уступая место обычному разграничению между достоверной и недостоверной работой. А биоинформатика перестает быть прикладной областью, лишь обслуживающей экспериментальные исследования, и превращается в самостоятельное научное направление, тесно связанное с современной биологией.В рамках этого направления решаются такие задачи, как функциональная аннотация отдельных генов и полных геномов, метаболическая реконструкция, анализ регуляторных систем, теория молекулярной эволюции в ее многообразных проявлениях - от эволюции отдельных генов и белков до эволюции метаболических путей, регуляторных систем и целых геномов, изучение статистических свойств геномных последовательностей, предсказание пространственной структуры биополимеров по последовательностям, реконструкция начальных этапов возникновения генетической информации. Помимо геномики, в научный обиход вошли такие термины, как протеоми-ка (анализ белков на уровне целого генома), транскриптомика (изучение экспрессии генов), метаболомика (анализ метаболизма путем одновременного измерения клеточных концентраций многих веществ). Начали говорить о наступлении постгеномной эпохи. Вычислительные методы стали не только составной компонентой любого массового исследования (поскольку без них невозможно осуществлять даже предварительную обработку и хранение данных), но и основным средством для получения из этих данных биологически содержательной информации.
К числу самых продвинутых направлений современной индустриальной биологии относится анализ экспрессии генов на олигонуклеотидных чипах [9]. Эта технология позволяет измерять концентрации матричной РНК (мРНК), соответствующие каждому гену организма, и выделять группы генов, одинаково реагирующих на изменение условий. Для таких генов можно предполагать одинаковую регуляцию, а потому следующим этапом становится выявление сигналов, которые отвечают за эту регуляцию. Проанализировав уровни экспрессии на последовательных стадиях какого-либо процесса, можно построить схему регуляторных взаимодействий и в конечном счете описывать генные сети, отвечающие за развитие этого процесса. Среди исследованных систем такого рода - клеточное деление у дрожжей, развитие цветка растений и зародыша дрозофилы [10].
Наконец, сравнение наборов генов, экспрессирующихся в нормальных или раковых клетках, а также в опухолях различной этиологии, позволяет выделять относительно небольшие группы генов, по которым опухоль отличается от нормы и разные виды опухолей различаются между собой. Таким образом создаются диагностические системы для дифференциальной диагностики. Их надежность часто оказывается выше, чем у традиционных цитологических и гистологических методов, поэтому применение таких систем может существенно изменить медицинскую практику, так как диагнозы станут точнее, а лечение - адекватное.
Анализ клонотек * интересен и сам по себе. В разных странах осуществляется несколько проектов по тотальному секвенированию мРНК человека, а значит, будет получена полная характеристика набора человеческих генов. Даже частичные результаты секвенирования, так называемые экспрессируемые ярлыки (Expressed Sequence Tags - EST), не только служат источником для идентификации генов, но и используются сами по себе, например, при изучении альтернативного сплайсинга. Именно анализ данных EST позволил оценить частоту альтернативного сплайсинга генов человека (более 50%) и показал, что альтернативный сплайсинг является одним из фундаментальных механизмов порождения белкового разнообразия, а не относительно редким вариантом генной регуляции, как думали раньше. Проведя сравнение геномов мыши и человека, мы установили, что более половины альтернативно сплайсируемых генов имеют видоспецифичные изоформы [11], так что, по всей вероятности, альтернативный сплайсинг - важный механизм эволюции эукариот.
* Набор клонированных фрагментов кДНК, полученных в одних условиях из одного источника.Анализ уровней экспрессии генов, в первую очередь, интересен потому, что гены кодируют белки. Однако данные об уровне транскрипции дают лишь приближенное представление о концентрации соответствующего белка из-за многих посттранскрипционных механизмов, регулирующих уровень трансляции, скорость деградации мРНК и белков, а также из-за разнообразных химических модификаций белковых молекул (фосфорилирование, гликозилирование и т.п.). Поэтому была поставлена задача дать полную характеристику всех белков, измерить их концентрацию в клетке, для чего совершенствуются технологии, основанные на использовании масс-спектрометрии [12]. Заметим, что в протеомике, как и в классической геномике, основной прием анализа масс-спектрометрических данных по-прежнему состоит в их сопоставлении с геномными последовательностями. Для этого разрабатываются специальные алгоритмы.Другой областью протемики стал массовый анализ пространственных структур белков. Уже довольно давно было замечено, что самый надежный метод предсказания пространственной структуры белка основан на сравнении его последовательности с последовательностями белков, структуры которых уже известны. Еще в конце 1990-х годов началось систематическое определение пространственных структур белков методами рентгеноструктурного анализа и ядерного магнитного резонанса. Подбор белков, структура которых должна быть установлена, также проводится с помощью методов сравнительной геномики. Прежде всего кластеризуют белки в родственные семейства так, чтобы структуры всех белков одного семейства были близки, затем выявляют семейства, не имеющие представителей с известной структурой, а уже потом рассматривают тот член семейства, который наиболее удобен для экспериментального анализа, то есть может быть выделен в достаточных количествах, хорошо кристаллизуется и т.п. Можно экспериментировать с несколькими белками, и даже если структуру удастся определить только у одного из них, этого достаточно для описания в целом структуры семейства.
Наконец, еще одно направление протеомики - массовый анализ белок-белковых взаимодействий. Для этого, во-первых, используются методы масс-спектрометрии, применяемые к белковым комплексам, например, к сплайсосомам. Во-вторых, если (опять же из анализа генома) установлен полный набор белков данного организма, то используется техника дрожжевых двугибридных систем; она также модифицирована для анализа взаимодействий белок - РНК и белок - лиганд.
При разработке проектов массовых исследований были учтены уроки, полученные в ходе выполнения программы "Геном человека". В частности, проводится большая работа по унификации данных об экспрессии генов и созданию специальных баз данных. Принято решение, что в процессе публикации результаты экспериментов по массовому анализу экспрессии генов на микрочипах должны быть помещены в соответствующий банк данных (подобно тому, как это делается с нуклеотидными последовательностями). Поскольку в формат базы данных заложены принципы контроля качества заполнения, можно надеяться, что удастся избежать загрязнения базы большим количеством недостоверных или просто плохо занесенных записей. Аналогичные базы данных разрабатываются и в рамках протеомных проектов. Таким образом, уже на стадии планирования проекта используются современные информационные технологии, учитывающие необходимость дальнейшего вычислительного анализа накапливаемой информации.
Сейчас идет работа над проектом по определению точечных полиморфизмов в геноме человека [13]. Она проводится под эгидой международного консорциума, основанного в 1999 г. несколькими фармацевтическими компаниями. Несмотря на коммерческое происхождение спонсоров проекта, все получаемые в нем данные доступны для любого анализа. К настоящему времени определены примерно 1800 тыс. полиморфизмов, причем для 100 тыс. из них известны аллельные частоты. Эти данные не только полезны для понимания эволюции человека, но и (что, наверное, важнее) являются неоценимым ресурсом медицинской генетики. С их помощью, как надеются исследователи, удастся картировать гены многофакторных наследственных заболеваний и гены индивидуальных предрасположенностей к болезням.
Продолжается и секвенирование геномов. Стоимость секвенирования все время падает, и потому поток полных геномов все нарастает. На разных стадиях находятся проекты по секвенированию геномов множества позвоночных, насекомых, паразитических червей, растений, грибов (в основном растительных и животных патогенов), простейших и, разумеется, бактерий. Более того, планируется тотальное секвенирование целых экологических ниш. Например, французский центр Genoscope, участвовавший в секвенирова-нии геномов человека, малярийного комара и многих бактерий, объявил о проекте Cloaca maximuma - тотального секвенирования бактериального сообщества сточных вод.
Заметим, что биологические задачи, о которых говорилось выше, часто приводят к сложным и пока далеким от эффективного решения математическим проблемам, в основном из области построения быстрых и эффективных алгоритмов для заведомо переборных задач. Возникающая принципиальная трудность может преодолеваться на пути уточнения постановки биологической задачи (экспериментальное определение ее существенных параметров, более глубокое понимание соответствующего ей биологического процесса), а также разработки математически обоснованных подходов к построению так называемых эмпирических алгоритмов, которые тоже могут иметь хорошее математическое основание. Уточнение и обсуждение списка биологических задач и соответствующих им математических проблем было бы полезно для развития биоинформатики.
Как известно, исследование генома прошло долгий путь развития и принесло много фундаментальных открытий (таблица). Биоинформатика делает первые шаги на пути превращения из инструмента хранения и обработки биологической информации в средство получения нового знания. В этот процесс вносят заметный вклад и отечественные специалисты, чему в немалой степени способствуют хорошие российские традиции в области информационных технологий и вычислительной молекулярной биологии.
Основные события в истории геномики
(нт - нуклеотид)
Год Автор Открытие, новая методика 1866
1869
1902
1910
1928
1938
1941
1944
1949
1953
1954
1955
1956
19571958
1959
1960
1961
1961
1962
1964-19661965
1967
1970
1972
1974-19791976
1975-1977
1977
1977
1977
1982
1983
1986
1986
1987
1988
1995
1996
1996
1996
1997
1997
1998
2000
2000
2000-2001
20022003
Г. Мендель
Ф. Мишер
А.Е. Гэррод
Т.Х. Морган
Ф. Гриффитс
Р. Сигнер
В. Эстбюрн
0. Эвери
Э. Чаргафф
Дж. Уотсон, Ф. Крик, М. Уилкинс, Р. Франклин
Г.А. Гамов
Ф. Сэнгер
В.М. Ингрэм
Ф. КрикМ. Мезельсон, Ф. Сталь
А. Корнберг
М. Перуц, Дж. Кендрю
Дж. Мармур, П. Доти
Ф. Жакоб, Ж. Моно
В. Арбер, X. Смит
Ф. Крик, М. Ниренберг, Дж.X. Маттеи,
С. Очоа, X. Г. Корана
Э. Цукеркандль, Л. Полинг
М. Геллерт
X. Темин, Д. Балтимор
В. Бойер, С. Коэн, П. Берг
С. Тонегава, Л. Худ
Ф. Сэнгер, Ф. Максам, В. Гилберт
Ф. Шарп, Р. Робертc
К. Вёзе
К. Муллис
Л. Худ, Т. Хункапиллер
DOE, NIH, Wellcome trust
Международный консорциум, Celera
Международный консорциум
Концепция гена и основы генетики
Выделение ДНК
Генетические болезни человека
Связь генов и хромосом
Трансформация бактерий
Молекулярная масса ДНК
Первая рентгенограмма ДНК
Трансформирующий фактор - это ДНК
Правила Чаргаффа - совпадение частот А-Т и G-C
Двойная спираль
Идея генетического кода
Аминокислотная последовательность инсулина
Молекулярная основа серповидноклеточной анемии
"Центральная догма" - перенос информации
от нуклеиновых кислот к белкам
Репликация ДНК на основе комплементарности нитей
ДНК-полимераза
Структуры гемоглобина и миоглобина
Ренатурация ДНК
Регуляция экспрессии генов
Рестриктазы
Расшифровка генетического кодаМолекулярные часы
ДНК-лигаза
Обратная транскриптаза
Клонирование ДНК
Порождение разнообразия иммуноглобулинов
путем рекомбинации
РНК-фаг MS2 (3569 нт*)
Методы быстрого определения нуклеотидной последовательности
ДНК-фаг (р XI 74 (5386 нт)
Сплайсинг
Архебактерии
Банки нуклеотидных последовательностей
ДНК-фаг Т7 (39936 нт)
Хлоропласт табака (155939 нт)
Полимеразная цепная реакция
Автоматический секвенатор
Начало проекта "Геном человека"
Геном бактерии Haemophilus influenzae (1830138 нт)
"Бермудские принципы" - немедленная публикация
секвенированных последовательностей
Секвенирование геномов:
архебактерия Methanococcus jannashii (1664970 нт)
дрожжи Saccharomyces cerevisiae (12 млн. нт)
Escherichia coli (4639221 нт)
Bacillus subtilis (4214814 нт)
нематода Caenorhabditis elegans (97 млн. нт)
растение Arabidopsis thaliana (115 млн. нт)
Drosophila melanogaster (120 нт)
черновик генома человека (3 млрд. нт)
мышь, рыба (фугу), малярийный комар и
малярийный плазмодий, рис
99.99% генома человека
ЛИТЕРАТУРА
1. Doolittle R.F., Hunkapiller M.W., Hood L.E. et al. Simian sarcoma virus oncogene, v-sis, is derived from the gene (or genes) encoding a platelet-derived growth factor//Science. 1983. V. 221. P. 275-277.
2. Lyubetsky VA., V'yugin V.V. Methods of horizontal gene transfer determination using phylogenetic data // In Silico Biology. 2003. № 3. P. 1-15.
3. Гельфанд М.С., Миронов А.А. Вычислительная биология на рубеже десятилетий // Молекулярная биология. 1999. Т. 33. С. 969-984.
4. Koonin E.V., Galperin M.Y. Sequence-Evolution-Function: Computational approachers in comparative genom-ics. Kluwer Academic Press, 2003.
5. Osterman A., Overbeek R. Missing genes in metabolic pathways: a comparative genomics approach // Curr. Opin. Chem. Biol. 2003. V. 7. P. 238-251.
6. Vitreschak А.А., Rodionov DA., Mironov А.А., Gelfand M.S. Regulation of riboflavin biosynthesis and transport genes in bacteria by transcriptional and transia-tional attenuation // Nucleic Acids Research. 2002. Т. 30. P. 3141-3151.
7. Winkler W.C., Cohen-Chalamish S., Breaker R.R. An mRNA structure that controls gene expression by binding FMN // Proc. Natl. Acad. Sci. USA. 2002. V. 99. P. 15908-15913.
8. Любецкая Е.В., Леонтьев Л.А., Гельфанд М.С., Любецкий В.А. Поиск альтернативных вторичных структур РНК, регулирующих экспрессию бактериальных генов // Молекулярная биология. 2003. Т. 37. № 5.
9. Yershov G., Barsky V., Belgovskiy A. et al. DNA analysis and diagnostics on oligonucleotide microchips // Proc. Natl. Acad. Sci. USA. 1996. V. 93. P. 4913-4918.
10. Ananko E.A., Podkolodny N.L., Stepanenko I.L. et al. CeneNet: a database on structure and functional organisation of gene networks // Nucleic Acids Res. 2002. V. 30. P. 398-401.
11. Nuritdinov R.N., Artamonova I.I., Mironov AA., Gelfand M.S. Low conservation of alternative splicing patterns in the human and mouse genomes // Hum. Mol. Genet. 2003. V. 12. P. 1313-1320.
12. Patterson S.D.,Aebersold R.H. Proteomics: the first decade and beyond // Nature Genetics. 2003. V. 33. P. 311-323.
13. Thorisson G.A., Stein L.D. The SNP Consortium web-site: past, present and future // Nucleic Acids Res. 2003. V. 31. P. 124-127.