№11, 2005 г.
© Ковальзон В.М.
Раскрыта природа нарколепсии В.М.Ковальзон
Владимир Матвеевич Ковальзон, доктор биологических наук,
ведущий научный сотрудник Ин-та проблем экологии и эволюции им.А.Н.Северцова РАН,В 1880 г. французский врач Эдуард Желино впервые описал тяжелое и неизлечимое неврологическое заболевание и назвал его нарколепсией. Для этой болезни характерны внезапные приступы катаплексии - полного расслабления скелетных мышц, когда человек не может ни пошевелиться, ни заговорить, хотя способность к непроизвольному дыханию сохраняется. Такой приступ продолжается обычно несколько минут (редко - до получаса) и провоцируется сильными раздражителями: возбуждением, смущением, страхом, гневом, физическими упражнениями, половыми сношениями, но особенно часто - хохотом, когда и у здорового-то человека, как говорится, “коленки подгибаются”.
В нашей стране это весьма редкое заболевание (возможно, просто потому, что его не умеют диагностировать); в Америке таких больных больше - около 125 тыс. человек, или 0.5-1 на 1000 жителей (лишь в четыре раза меньше, чем “паркинсоников”); больше всего их в Японии - 1 на 600 человек, а меньше - в Израиле: несколько человек на всю страну, или 1 на 500 тыс. жителей, т.е. в 100 раз меньше, чем в Японии! Первые симптомы болезни появляются в подростковом или юношеском возрасте, нарастают в течение года или двух и затем стабилизируются.
Вот уже лет сорок как нарколепсия привлекает особое внимание сомнологов - не только клиницистов, но и ученых. В одной из наших предыдущих публикаций мы рассказывали о результатах прямого изучения нейронов головного мозга, вовлеченных в регуляцию сна-бодрствования [1]. Тогда было обнаружено, что нормальная работа коры больших полушарий (а точнее, таламо-кортикальной системы мозга), обеспечивающих весь спектр сознательной деятельности человека, возможна только при наличии постоянных (тонических) мощных воздействий со стороны определенных подкорковых структур, называемых активирующими. Благодаря этим воздействиям мембрана большинства кортикальных нейронов во время бодрствования деполяризована на 5-15 мВ по сравнению с потенциалом покоя (–65/–70 мВ). Только в таком состоянии тонической деполяризации нейроны способны обрабатывать информацию и отвечать на сигналы, приходящие к ним от других нервных клеток, как рецепторных, так и внутримозговых.
Сегодня известно, что систем тонической деполяризации, или активации коры мозга (их можно условно назвать “центрами бодрствования”), несколько - вероятно, около десятка. Располагаются они на всех уровнях мозговой оси: в продолговатом мозге, в ретикулярной формации моста, среднего и межуточного мозга, в области синего пятна и дорзальных ядер шва, в заднем гипоталамусе и базальных ядрах переднего мозга. В качестве медиаторов нейроны этих отделов мозга выделяют глютаминовую кислоту, ацетилхолин, норадреналин, серотонин и гистамин (рис.1). Активность их модулируют многочисленные пептиды, находящиеся в тех же самых синаптических пузырьках - везикулах. У человека нарушение деятельности любой из этих систем не компенсируется за счет других, что несовместимо с сознанием и приводит к коме.
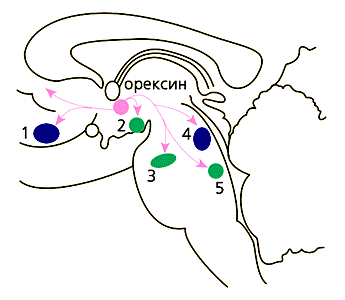
Рис.1. Схема расположения “центров бодрствования” в головном мозге человека
и влияния на них орексиновых нейронов (показано красным).
1 - базальные ядра переднего мозга (выделяют ацетилхолин),
2 - ядра заднего гипоталамуса (выделяют гистамин),
3 - дорзальные ядра шва (выделяют серотонин),
4 - область покрышки моста (выделяет ацетилхолин),
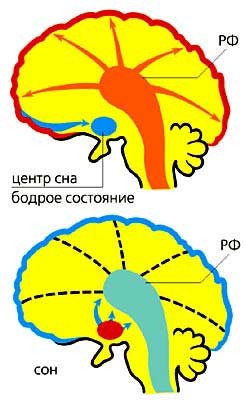
5 - синее пятно (выделяет норадреналин).Если есть “центры бодрствования”, то должны быть и “центры сна”, споры о которых велись еще со времен И.П.Павлова. В конце 80-х годов XX в. был, наконец, найден центр обычного (медленного) сна (рис.2). Выяснилось, что нейроны, активность которых незначительна в бодрствовании, но резко возрастает в период обычного сна и прекращается во время парадоксального, расположены в переднем гипоталамусе, в так называемом вентро-латеральном преоптическом ядре, а их медиатор - гамма-аминомасляная кислота (ГАМК) - главный тормозный медиатор головного мозга. Интересно, что мучительная бессонница, возникавшая у больных вирусным энцефалитом, эпидемия которого возникла во время Первой мировой войны, связана с поражением именно этого ядра. Этот синдром описал знаменитый австрийский невролог фон Экономо, который первый предположил существование “центра сна” в переднем гипоталамусе.
Рис.2. Схема расположения центра медленного сна. Вверху - бодрствование, когда центр сна заторможен и ретикулярная формация РФ (центр бодрствования) активирует кору;
внизу - центр сна возбужден, РФ заторможена и кора не активируется.
Как уже отмечалось в предыдущей статье, практически с момента открытия парадоксального (быстрого) сна стала ясна его ярко выраженная активная природа. Запускается он из четко очерченного центра, расположенного в эволюционно древней, задней части мозга, в области так называемого варолиева моста. Химическим передатчиком сигналов этих клеток служит в первую очередь ацетилхолин, а также глутамат. Хотя во время парадоксального сна клетки мозга чрезвычайно активны, но информация от “входов” (органов чувств) к ним не поступает и на “выходы” (мышечную систему) не подается. В этом и заключается парадоксальность такого состояния, отраженная в его названии. Видимо, в период быстрого сна интенсивно перерабатывается информация, полученная в предшествующем бодрствовании и хранящаяся в памяти. Подтверждением тому служат эмоционально окрашенные сновидения, появляющиеся у человека в парадоксальном сне.
Нарушения работы “центров сна” наблюдаются при некоторых довольно редких видах патологии, в том числе и при нарколепсии. У разных нарколептиков приступы отличаются некоторым своеобразием: одни сохраняют полный контакт с окружающей средой - все слышат, все чувствуют, все помнят, но не могут реагировать до окончания приступа; у других происходит “отключение” от внешнего мира и возникают так называемые гипнагогические галлюцинации - состояние, которое здоровые люди переживают во время засыпания и дремоты; третьи переживают яркие эмоциональные сновидения… Большинство нарколептиков плохо спит ночью; ночной сон у них раздроблен, фрагментарен, а дельта-сон (глубокие стадии медленного сна) и парадоксальный сон представлены недостаточно. Соответственно, такие больные испытывают постоянную очень сильную сонливость днем. Естественно, что им категорически запрещается водить машину; есть и другие ограничения в их профессиональной деятельности. От нарколепсии не умирают, но жизнь превращается в сплошное мучение…
До конца минувшего века нарколепсия оставалась загадочным заболеванием. Какие только факторы не предлагались врачами в качестве ее причины! Одни неврологи относили ее к психосоматическим заболеваниям; другие считали одним из проявлений шизофрении; третьи полагали, что она есть следствие нарушения нейрохимического равновесия в головном мозге; четвертые выдвигали гипотезу о ее вирусном происхождении; а один немецкий психоневролог даже предположил, что нарколепсию вызывает чрезмерная мастурбация в подростковом возрасте! Очевидно было одно - в формировании нарколепсии играют роль как некие врожденные факторы, так и какие-то внешние воздействия.
Предположили, что болезнь специфически связана с “поломкой” системы, запускающей парадоксальную фазу сна. В экспериментах на кошках ряд исследователей, в том числе крупнейший сомнолог Мишель Жуве с сотрудниками, показали, что отдельные проявления парадоксального сна (мышечное расслабление и подергивание, быстрые движения глаз, характерные изменения электрической активности мозга и пр.) вызваны активностью отдельных, пространственно разделенных групп нервных клеток в “центре парадоксального сна”. Напомним, что они находятся в древних, задних отделах мозга (латеродорзальной/педункулопонтинной области покрышки моста) и выделяют в качестве медиатора (нейропередатчика) молекулы ацетилхолина. Детальное изучение нарколептических приступов показало, что в большинстве случаев они представляют собой не что иное, как внезапное, совершенно неадекватное включение механизма парадоксального сна прямо из бодрствования! А разнообразие форм приступов отражает преимущественное поражение тех или иных отдельных частей этого механизма.
Для понимания сути таких нарушений нужна была экспериментальная модель нарколепсии. Обнаружили ее случайно: однажды в середине 60-х годов знаменитый американский сомнолог Вильям Демент, один из первооткрывателей парадоксального (быстрого) сна, ученик легендарного Натаниэля Клейтмана, как-то рассказывал друзьям о пациентах-нарколептиках, с которыми тогда работал в клинике. Вдруг один из знакомых воскликнул: “Позволь, но ведь то, что ты так красочно описываешь, очень похоже на приступы, которые я иногда наблюдаю у своего добермана!”.
Действительно, из ветеринарной и кинологической литературы выяснилось, что у домашних животных - собак, коров и лошадей - изредка отмечаются катаплексические приступы, похожие на нарколепсию человека. У собак эти приступы также провоцируются эмоциональным возбуждением, происходящим во время игры или, чаще всего, при виде любимой пищи. Заболевание наследуется по так называемому аутосомно-рецессивному типу; значит, для получения стопроцентно больного потомства необходимо, чтобы “нарколептиками” были оба родителя. Демент и его сотрудники с помощью классических методов селекции (скрещивания и отбора) вывели “чистую линию” собак-нарколептиков - доберман-пинчеров и лабрадоров. Вскоре их была уже целая стая.
Изучение нарколепсии у собак продолжалось около 20 лет и принесло немало интересных результатов. К тому времени было известно, что в самой задней (каудальной) части мозга - продолговатом мозгу - есть особая группа нейронов, посылающих свои длинные отростки, аксоны, в спинной мозг, для осуществления функции торможения произвольных движений. Основной тормозной медиатор этой системы - аминокислота глицин. Когда человек или животное двигается, эти нейроны почти не разряжаются, в покое и во сне усиливают частоту разрядов, а в парадоксальном сне, когда появляются сновидения и мышечная деятельность полностью подавлена, они максимально активны. Если какая-то часть системы разрушена - будь то в эксперименте (у крыс, кошек и собак) или в результате заболевания (у человека), то во время парадоксального сна возникают движения, соответствующие переживаемому сновидению… Так вот, при приступах катаплексии, как и в парадоксальном сне, у собак отмечались мощные вспышки активности этих нейронов!
Другая группа нейронов, расположенная несколько выше по мозговой оси и образующая так называемое “синее (или голубое) пятно”, знаменита тем, что там синтезируется почти весь норадреналин мозга - один из важнейших “медиаторов бодрствования”.
Норадренергических нейронов в мозге мало, но благодаря своим мощным “древовидным” ветвлениям они иннервируют огромное количество других нейронов во всех частях мозга. У нейронов синего пятна обнаружили точно такие же эффекты, хотя и противоположные по знаку. Они, как и другие аминергические нейроны в “центрах бодрствования” (серотонинергические в дорзальных ядрах шва и гистаминергические - в туберомаммиллярном ядре заднего гипоталамуса) проявляли максимальную активность во время бодрствования, снижали ее во сне и полностью “замолкали” во время сновидений (парадоксального сна).
У собак во время нарколептических приступов эти нейроны вели себя так же, как при парадоксальном сне, т.е. тормозились. Активность серотонинергических нейронов при этом не прекращалась полностью, но сильно ослабевала - примерно как при обычном (медленном) сне, а гистаминергических - резко возрастала, почти до уровня активного бодрствования (рис.3)! Стало ясно, что норадренергические и, отчасти, серотонинергические нейроны ответственны за подавление мышечного тонуса, а гистаминергические - за сохранение сознания при катаплексии (собаки в этом состоянии способны следить глазами за перемещениями объектов в поле зрения). Значит, прекращение активности возбуждающих норадренергических нейронов и ее усиление у тормозных глицинергических и ГАМК-ергических - именно тот механизм, который снимает мышечный тонус во время парадоксального сна и приступов нарколепсии.
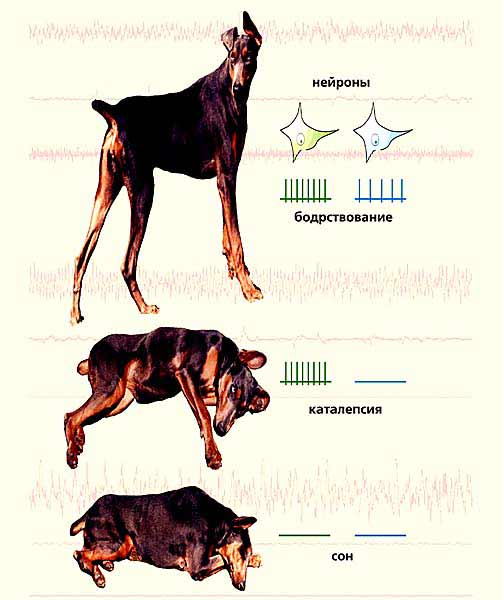
Рис.3. Электроэнцефалограммы головного мозга собаки-нарколептика.
Вверху - во время бодрствования; в середине - в приступе катаплексии; внизу - в состоянии обычного (медленного) сна. Розовым показана электрическая активность мозга (ЭЭГ, верхняя кривая), глаз (ЭОГ, средняя кривая) и мышц шеи (ЭМГ, нижняя кривая); внизу - только ЭЭГ и ЭМГ. Справа показано, как работают при этом одиночные нейроны - гистаминергический (зеленым) и норадренергический (синим).Так было совершенно определенно показано, что приступы нарколепсии со всеми присущими им проявлениями есть, по сути, не что иное, как “пароксизмы”, т.е. внезапные неадекватные включения нейронного центра парадоксального сна во время бодрствования. Иначе говоря, в нашем мозгу есть тормозной механизм, который не позволяет нам во время бодрствования разом “отключаться” от внешнего мира и видеть сны. Поломка этого механизма по каким-то причинам и приводит к нарколептическим приступам… Однако в чем именно она заключается - оставалось неизвестным.Со времен Желино патологоанатомы искали изменения в головном мозгу нарколептиков, которые можно было бы связать с развитием этого заболевания. Однако столетние усилия оставались тщетными. В последние 20 лет американские исследователи (Билл Демент, Кристиан Гийемино и Эммануэль Миньё) и французские специалисты из лаборатории Демента в Лос-Анджелесе, а также группа Томаса Килдаффа в Стенфордском университете (Сан-Диего) обнаружили повышенное содержание основных нейропередатчиков и их рецепторов в различных отделах головного мозга у собак-нарколептиков. Наконец, в начале 90-х годов подобные же биохимические и морфологические изменения нашли и в мозге больных людей при посмертном вскрытии…
Рецепторные белки представляют собой “молекулярные замочки”, расположенные на клеточной поверхности. А ключи к этим замочкам - нейропередатчики (медиаторы), называемые лигандами. Если ключ подходит (т.е. происходит лиганд-рецепторное взаимодействие), то либо резко меняется состояние клеточной мембраны - открываются одни и закрываются другие каналы, регулирующие потоки ионов (Na+, K++, Ca++, Cl-) внутрь и наружу клетки, либо изменяется состояние ее генетического аппарата - усиливается или ослабевает синтез какого-то специфического белка. Соответственно меняется способность нейрона отвечать на сигналы, поступающие от других клеток: они посылают свои импульсы третьим клеткам.
Служат ли выявленные изменения причиной или следствием заболевания? Невозможно было ответить на эти вопросы, пока в нейробиологии не произошла революция, связанная с появлением молекулярно-генетических методов. После публикации частично раскрытого генома собаки Миньё и его сотрудники сумели найти в одной из хромосом мутацию, предположительно ответственную за нарколепсию [2]. Однако и это еще не раскрывало ее механизм. Разгадка, как всегда, пришла неожиданно и стала последней сенсацией в ряду выдающихся открытий нейробиологии XX в.
В 1998 г. большая группа авторов, первые среди которых Луис Делиси и уже упоминавшийся Том Килдафф, опубликовала статью о присутствии в гипоталамусе матричной РНК (мРНК), кодирующей белок, содержащий два сходных пептида с неизвестными ранее аминокислотными последовательностями [3]. Их назвали гипокретинами (ГИПОталамическими сеКРЕТИНАМИ), так как поначалу приняли за представителей группы кишечных гормонов секретинов (в дальнейшем это сходство опровергли). Одновременно группа японских исследователей из Техасского университета, занимавшаяся поиском лигандов к “сиротским” рецепторам (т.е. “подбором ключей к найденным замкам”), нашла в гипоталамусе два близких по строению пептида и назвала их орексинами А и Б (от греч. orex - аппетит). Оказалось, что гипокретины 1 и 2 и орексины А и Б - одни и те же вещества. Были обнаружены два рецептора к орексинам/гипокретинам и кодирующие их гены. Рецептор 1-го типа связывается избирательно с орексином-А, а 2-го - с обоими пептидами. Вначале считали, что эти пептиды играют важную роль в регуляции пищевого поведения, так как тела нейронов, аксоны которых выделяют орексин/гипокретин, располагаются в глубине головного мозга, вблизи так называемого “пищевого центра”. Там находятся клетки, участвующие в регуляции голода и насыщения. Однако сейчас эта роль поставлена под сомнение [4].
Орексины-гипокретины представляют собой олигопептиды; в орексине А - 33 аминокислотных остатка, а в орексине Б - 28. У первого (А) свернутая (петлеобразная) пространственная структура, которая удерживается дисульфидными мостиками. Второй (Б) имеет линейную структуру. Оба пептида образуются в организме в результате расщепления одного белка-предшественника, пре-прогипокретина. Подобно аминергическим нейронам (например, клеткам синего пятна), орексинергические нейроны весьма немногочисленны (в полутораграммовом мозге крысы их всего 1700, а в тысячу раз большем по весу мозге человека - около 80 тыс.). Однако их длинные отростки - аксоны - сильно ветвятся, иннервируя множество клеток в самых разных отделах мозга, выделяющих все основные медиаторы: ацетилхолин, глутамат, ГАМК, мозговые амины… Интересно, что с ГАМК-ергическими нейронами “центра медленного сна” в вентролатеральном преоптическом ядре, расположенном в непосредственной близости от орексиновых нейронов латерального и вентро-медиального гипоталамуса, орексиновые нейроны оказались совсем не связаны [5]!
В большинстве нейронов орексин присутствует вместе с другим пептидом - динорфином. Во всех орексиновых клетках гипоталамуса имеется особый секретируемый нейрональный белок NARP (“пентраксин, регулирующий нейронную активность”). Он связан с регуляцией синапсов и кодируется одним из “немедленных ранних генов”, которые быстро экспрессируются (активируются) в нейронах при повышении их метаболической активности. В числе прочих орексинергические нейроны проецируются и на норадренергические клетки синего пятна, вызывая их деполяризацию - активацию, “подбуживание”. При недостаточной активации нейроны синего пятна могут внезапно “замолкать” не только во время парадоксального сна (как им положено), но и при бодрствовании, вызывая приступы нарколепсии [6].
Миньё и его команда обнаружили, что у собак из-за мутации гена, кодирующего один из двух рецепторов орексина, молекула белка NARP теряет способность связываться с своим лигандом. В это же время группа исследователей из Медицинского института Говарда Хьюза при Техасском университете в Далласе, используя опубликованную карту генома мышей, сумела вывести линию, у которой с рецепторами орексина было все в порядке, но сам лиганд - орексин - не выделялся. Эти искусственно выведенные мыши также послужили моделью нарколепсии - у них отмечались сходные явления.
Хотя проявления нарколепсии у человека и у собаки в целом почти тождественны, в отношении сходства их механизмов возникли серьезные сомнения. Достаточно сказать, что “собачья нарколепсия” - чисто генетическое заболевание, а человеческая - отнюдь нет! Она не передается по наследству, ее нельзя назвать семейным или родственным заболеванием. Если один из однояйцовых близнецов страдает нарколепсией, то у второго подобная болезнь наблюдается лишь в 25% случаев! Значит, у человека, в отличие от собаки, наследственность лишь определяет некоторую предрасположенность к болезни, но вовсе ее не гарантирует [7]. Следовательно, имеется еще какой-то ключевой фактор, не связанный с генетическими явлениями, разрушающий орексиновую систему у человека и таким образом имитирующий вышеописанные мутации у животных.
В 1999 и 2000 гг. появились сообщения о том, что нарколепсия, возможно, аутоиммунное заболевание, подобное хорошо известному паркинсонизму, при котором иммунная система ошибочно атакует некоторые клетки мозга, воспринимая их как чужеродные. Еще в 1984 г. выяснилось, что у 90% нарколептиков имеется особая разновидность группы генов лейкоцитарного антигена человека (HLA), а в целом у населения эта разновидность встречается лишь в 25% случаев. Белки, кодируемые генами этой области, участвуют в предъявлении антигенов клеткам иммунной системы, и многие иммунные заболевания связаны с одной или несколькими разновидностями (гаплотипами) группы HLA.
Посмертное изучение мозга нарколептиков с помощью новейших иммуногистохимических методов антигенного восстановления, проведенное в лаборатории известного сомнолога Джерома Зигеля из Лос-Анджелеса, выявило катастрофическую потерю 90% орексинергических нейронов и их аксонов, особенно в тех областях, где наблюдалась высокая насыщенность рецепторов 2-го типа. Анализ крови нарколептиков также показал, что концентрация орексина у них ниже уровня чувствительности метода (0,5 пг/мл), тогда как у здоровых людей в бодрствовании она составляет 50 пг/мл. Стало ясно, что, в отличие от экспериментальной нарколепсии у собак, вызываемой определенной точечной мутацией гена рецептора орексина 2-го типа, у человека нарколепсия - аутоиммунное заболевание [8]. Но какое вещество служит тем антигеном, против которого “восстает” иммунная система мозга человека, попутно поражая орексиновые клетки - остается неизвестным. Однако эта задача уже не нейрофизиологии, нейрохимии и нейроанатомии, а нейроиммунологии. Представители первых трех наук в ближайшие годы, видимо, продолжат изучение орексиновой системы “по вертикали и горизонтали”, т.е. ее формирование в ходе онтогенеза (индивидуального развития) и сравнительное ее изучение у животных, стоящих на разных уровнях развития, для реконструкции возможного хода эволюции этой системы мозга.
Открытие орексиновой системы - крупнейшее достижение нейробиологии, которым она завершила XX век, столь богатый для нее революционными успехами. Природа нарколепсии, болезни, остававшейся загадкой на протяжении 120 лет, было раскрыта всего за 2.5 года, продемонстрировав всю мощь и гибкость современной науки. Эта история еще раз (уже в который!) доказывает, что самые, казалось бы, далекие от практических задач работы в фундаментальной науке способствуют значительным достижениям в медицине: раскрытию истинных причин тяжелых заболеваний, новым методам диагностики и лечения, принципиально новым лекарственным средствам. И наоборот, “экономия” на фундаментальных исследованиях, которые кажутся невеждам слишком “абстрактными”, приводит к полному прекращению всякого прогресса в прикладных исследованиях, к их полной остановке “в бессильи умственного тупика”…
Литература
1. Ковальзон В.М. Природа сна // Природа. 1999. №8. С.172-179.
2. Mignot E. // Sleep Medicine. 2000. V.1. №1. P.87-90.
3. Kilduff T.S., Peyron C. //TINS. 2000. V.23. №8. P.359-365.
4. John J. et al. // Neuron. 2004. V.42. №5. P.619-634.
5. Scammel T.E. // Current Biology. 2001. V.11. P.R769-R771.
6. Ferguson A.V., Samson W.K. // Frontiers in Neuroendocrinology. 2003. V.24. №1. P.141-150.
7. Siegel J.M. // Annual Review of Psychology. 2004. V.55. №2. P.125-148.
8. Siegel J.M. et al. // Neuropsychopharmacology. 2001. V.25. №S5. P.S14-S20.