№ 4, 2003 г.
© Д.А. Леменовский, Д.П. Крутько, М.В. Борзов
Циркониевые сандвичи
для активации углеводородовД.А.Леменовский, Д.П.Крутько, М.В.Борзов
Дмитрий Анатольевич Леменовский, д. х. н., профессор,
заведующий лабораторией координационных металлоорганических соединений
химического факультета МГУ им.М.В.Ломоносова.Дмитрий Петрович Крутько, к. х. н., с. н. с. той же лаборатории.
Максим Владимирович Борзов, к. х. н., в. н. с. той же лаборатории.
В начале всяческой философии лежит удивление, ее развитием является исследование, ее концом — незнание. М.Монтень
По существу каждое исследование задает вопросы природе и заканчивается не только готовыми результатами, но и появлением новых вопросов. Вероятно, именно в таком смысле следует понимать слово “незнание” в эпиграфе.
Основой промышленного синтеза громадного числа органических соединений и производства полимеров служит главный сырьевой источник — нефть. Не менее значим также природный газ, использование которого для органического синтеза предпочтительнее, но пока менее развито. К сожалению, природа заготовила для нас запасы химически связанного углерода не в самом удобном виде. Известно, что чем инертнее вещество, тем оно лучше сохраняется, и именно потому существуют его большие запасы. Почти на 80% нефть состоит из насыщенных углеводородов — парафинов (их название указывает на химическую инертность: лат. parum affinis означает лишенный сродства) и нафтенов (алициклических углеводородов).
Если мы хотим использовать нефть и газ как сырье для синтеза, то должны превратить исходные углеводороды в соединения с функциональными, реакционноспособными, группами. Для этого необходимо разорвать С–Н- либо С–С-связи. Сегодня довольно хорошо разработаны методы таких превращений, однако большинство из них, такие как, например, крекинг (разрыв связи С–С) или галогенирование (разрыв связи С–Н), очень энергоемки и не селективны, т.е. дают много различных продуктов. Итак, существует потребность в избирательной переработке углеводородов, чтобы получать полезные продукты. Для этого применяют катализ.
Чем он привлекателен?
Известно, что стабильные химические вещества инертны потому, что располагают низким запасом энергии, и для их превращения в другие соединения необходимо преодолеть большой энергетический барьер, высоту которого измеряют энергией активации. Барьер можно преодолеть, существенно повысив температуру или давление, либо другим способом ужесточив условия. В результате получим скорее всего не только нужный продукт, но и “осколки” соединений, участвующих в реакции. В присутствии катализатора энергия активации бывает намного ниже, и реакция может пойти по иному пути. Кроме того, переходный комплекс катализатора с реагентом, имеющий строго определенное строение, способен придать реакции высокую избирательность.
Катализ в химии можно уверенно сравнить с эндохирургическими методами, которые позволяют проводить операцию, не вскрывая брюшную полость, а подводя инструмент к нужному месту по кровеносным сосудам.
Современная научная мысль направлена на поиск катализаторов нового типа, приближающихся по свойствам к тем, что существуют в живой природе. Например, известны бактерии, которые усваивают атмосферный азот или парафиновые углеводороды, причем исключительно деликатно, при обычных температуре и давлении. Исследования показали, что во всех подобных случаях в роли катализаторов выступают комплексные соединения металлов. Разрабатывая новые катализаторы, химики не всегда стараются копировать природу, чаще стремятся найти другие, не менее эффективные системы, которые позволяют не только получить нужный результат, но и понять сам механизм происходящих превращений. А это дает в руки исследователя способ управлять свойствами будущего катализатора.
Раздумья перед походом
Возможность каталитического расщепления С–Н-связи при участии комплексов металлов впервые обнаружили Дж.Чатт и Дж.Дэвидсон в 1965 г. Получив фосфиновый комплекс рутения, они установили, что соединение может перегруппировываться, при этом атом рутения внедряется по связи С–Н в метильной группе лиганда: [L(CH3)]2—Ru ® ® L(CH3)—Ru(H)–CH2L (L — фосфиновый лиганд). Вслед за тем усилиями многих исследователей удалось установить, что некоторые комплексы иридия и платины тоже способны разрывать связь С–Н [1], причем не только в органической группе лиганда, но и в насыщенных углеводородах, специально введенных в реакционную систему.
До определенного момента полагали, что справляться со столь трудной задачей могут только соединения благородных металлов. Однако интенсивные исследования комплексов переходных металлов косвенно указывали на то, что катализировать разрыв связи С–Н должны также соединения “ранних” переходных элементов (т.е. тех, что стоят в начале их ряда в каждом периоде), но лишь в том случае, если они находятся в низкой степени окисления. В первую очередь интересны в этом отношении титан, цирконий и гафний. Основная трудность при использовании таких элементов состоит в том, что в низких степенях окисления они крайне не стабильны. Поэтому прежде всего предстояло найти способ их защиты от возможного окисления.
Но вначале нужно было решить, соединения какого типа следует предпочесть. Опыт работы с благородными металлами не мог быть полезен, поскольку у элементов IV группы химические свойства несколько иные. В качестве основы были выбраны p-комплексы переходных металлов, прямые “потомки” ферроцена — знаменитого соединения 20-го столетия [2]. Эти комплексы имеют сандвичевое строение: в них атом металла зажат между двумя плоскими органическими циклами, в нашем случае пентадиенильными. Химия таких циклических соединений на сегодня хорошо разработана и открывает исключительные возможности для конструирования разнообразных комплексов.
Выбор металла мы сделали в пользу циркония, поскольку именно от него можно было ожидать более высокую активность. Впрочем, это не означает, что титан и гафний в той же роли менее интересны. Перевести атом металла, находящийся в структуре комплекса, из высшей степени окисления в низшую сравнительно несложно, но необходимо защитить его от окисления. Проще всего это сделать с помощью координирующих лигандов, а чтобы они в дальнейших реакциях не могли далеко отойти от комплекса, необходимо их прикрепить каким-либо образом к металлсодержащей молекуле. Таким требованиям полностью отвечают так называемые хелатные (chelat — клешня) лиганды. Они одним концом химически связаны с циклопентадиенильным фрагментом, а другим — с координирующим атомом (склонным к образованию донорно-акцепторной связи с металлом), в первую очередь из числа непереходных элементов V и VI групп, т.е. азотом, кислородом, фосфором или серой.
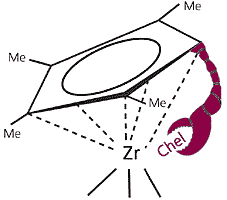
Длина “клешни” должна быть такой, чтобы группа, содержащая хелатирующий атом, легко дотягивалась до металла. Ее можно определить расчетом или (что гораздо интереснее) с помощью структурных моделей. Мы выбрали цепочку из двух метиленовых групп –CH2–CH2–. Помимо хелатной защиты металлического центра от окисления мы дополнительно экранировали молекулу, присоединив к пентадиенильному кольцу “торчащие усы” метильных групп (рис.1). Таким образом, общая схема была намечена, осталось лишь ее реализовать.
Рис.1. Модель метилированного циклопентадиенильного лиганда
с “клешней” (Chel).Долгой лестницы ступени
Неизбежная и весьма трудоемкая часть нашего плана — получение необходимых лигандов, содержащих хелатирующий хвост. Кроме того, необходимо было заранее предусмотреть, чтобы такие лиганды могли образовывать p-комплексы с металлом.
Мы обратились к мощному аппарату классической органической химии. В последние годы ее явно затмевают металлоорганическая химия и биохимия, однако, именно искусство органического синтеза, созданное нашими предшественниками, позволило двум указанным дисциплинам достичь современных высот.
Мы не можем отказать себе в удовольствии показать хотя бы в общих чертах всю цепочку синтеза таких лигандов [3, 4].
Вначале рассмотрим получение лигандов с хелатирующей группой PR2 (рис.2). На первой стадии метилированный циклопентадиен образует с литием ионное соединение, которое далее при взаимодействии с эфиром толуолсульфокислоты (тозилатом, Ts) переходит в соединение, содержащее ветку –CH2–CH2–Cl, и представляет собой смесь изомеров. Об этом можно судить по тому, что у одного атома водорода связь направлена не к конкретному углероду, а в середину цикла. Так что изомеры отличаются положением атома водорода — он может занимать место у любого углеродного атома в цикле. Все изомеры находятся в равновесии, но если один из них начнет участвовать в какой-либо реакции, его концентрация понизится и оставшиеся изомеры станут перестраиваться, чтобы повысить до равновесного уровня количество исчезающего изомера. Тот изомер, у которого блуждающий атом водорода находится в точке присоединения ветви –CH2–CH2–Cl, отщепит HCl, и в результате возникнет спироцикл (два цикла, соединенные одним общим атомом). На последней стадии трехчленный цикл размыкается под действием диметилфосфида (или дифенилфосфида) лития R2PLi. В итоге образуется запланированный лиганд: метилированный циклопентадиенильный анион с хелатным хвостом –CH2–CH2–PR2. И кроме того — катион лития, который позволит в дальнейшем присоединить к лиганду цирконий.
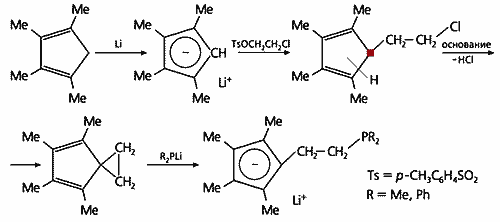
Рис.2. Получение лиганда с фосфидной хелатирующей группой.
Для получения лигандов, содержащих хелатирующие атомы азота, кислорода или серы, рассмотренный способ не применим, поскольку никакие реагенты с этими атомами не способны раскрывать спироцикл. Поэтому мы решили собирать пентадиенильные циклы из фрагментов, поместив хелатирующие ветви в исходные заготовки (рис.3). От бромированного бутена переходим к его литийпроизводному. Две такие молекулы объединяем в одну с помощью связывающего центра, который уже содержит ветку с хелатирующей группой (это было предусмотрено заранее). Разветвленную молекулу замыкаем в цикл и далее переводим его в литийпроизводное. В результате получаем такие же лиганды, как тот, что показан на рис.2, но с иными хелатирующими атомами.
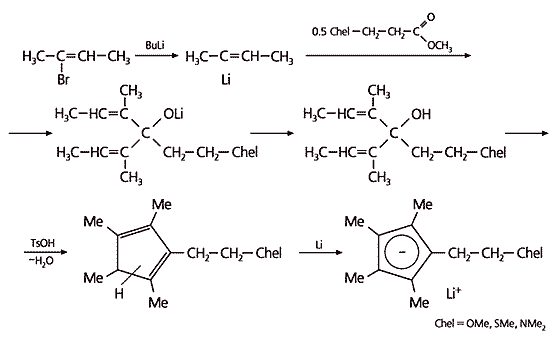
Рис.3. Синтез лигандов с хелатирующими атомами кислорода, серы или азота.
Теперь можно приступать к синтезу металлсодержащих комплексов. Общая схема присоединения лиганда к атому металла традиционная — взаимодействие литийпроизводного циклопентадиена (этот лиганд обозначен в схеме как ЦП) с галогенидом металла:
[ЦП]–Li+ + ClM– ® ® ЦП- - - - М + LiCl.
Именно такая простая на первый взгляд реакция приводит к сандвичевым соединениям, где атом металла связан не с конкретным углеродным атомом лиганда, а со всем циклом сразу, в данном случае все определяет химическая природа ЦП-лиганда.
Так как мы двигались к новым соединениям, то не могли заранее сказать, насколько плотно следует укрывать атом циркония, вполне возможно, что и одной хелатирующей группы достаточно, но мы запланировали различные варианты [5, 6]. Наши исходные соединения позволяют получить несколько структурных типов комплексов: полусандвич (рис.4, структура 1), а также сандвичи с двумя или одной хелатирующей веткой (структуры 2 и 3).
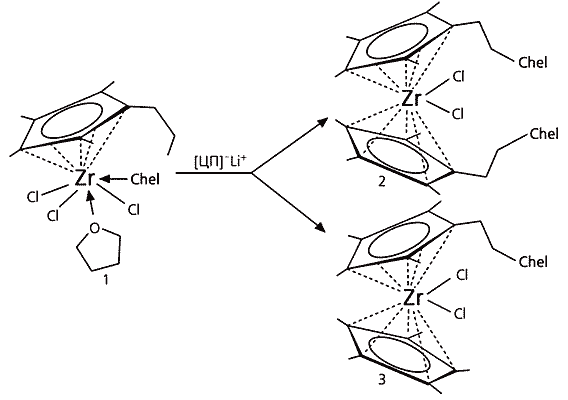
Рис.4. Циркониевые комплексы — полусандвич и два сандвича.
Метильные группы у ЦП-лиганда обозначены упрощенно, в виде валентных черточек.Не будем забывать, что мы имеем четыре типа хелатирующих веток, следовательно, возможны 12 вариантов комплексов. На самом деле их больше, так как у хелатирующего атома могут располагаться различные органические группы (у нас метильная Ме или фенильная Ph). Кроме того, в процессе синтеза, как выяснилось, могут появиться различные модификации, например те, что содержат молекулы сольватированного растворителя.
Итак, мы предложили цирконию разнообразное облачение. Теперь нужно было проследить за его поведением в комплексах. Рассмотрим лишь некоторые из них.
Цирконий сразу обнаружил особенности своего характера. Во всех полусандвичах он отчетливо демонстрировал координационную ненасыщенность, привлекая для заполнения координационной сферы не только хелатную группу, но и растворитель, например тетрагидрофуран. Молекулу растворителя цирконий охотно обменивал на ЦП-лиганд с хелатирующей веткой или без нее, отчетливо показывая, что с ЦП-лигандом возникают энергетически более выгодные структуры.
В полностью завершенных сандвичах (с двумя ЦП-лигандами) цирконий чувствовал себя вполне комфортно и не проявлял интереса к хелатирующим группам. Это позволило нам скорректировать дальнейшие действия и использовать для последующего восстановления только сандвичи.
Радуга превращений
Самый интересный этап — восстановление полученных комплексов [5, 6]. Восстанавливающим реагентом была амальгама магния, которая легко забирает атомы Cl от Zr(IV), превращая его в Zr(II). Забегая вперед, скажем, что цирконий полностью оправдал наш интерес к нему, а в некоторых случаях сумел удивить спектром своих превращений (рис.5).
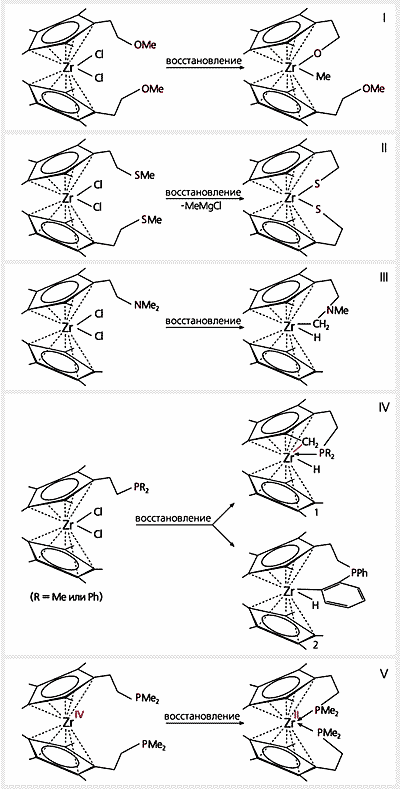
Рис.5. Восстановление амальгамой магния комплексов циркония,
содержащих разные хелатирующие группы.Одно свойство циркония проявилось сразу: образующийся при восстановлении Zr(II) крайне неустойчив, стремится перейти в исходное состояние Zr(IV) и использует для этого все, что находится рядом, в первую очередь — хелатирующие группы (Chel). В случае, когда Chel = ОМе, цирконий отрывает метильную группу от атома кислорода и присоединяет их по отдельности (рис.5, схема I). Все происходит точно так же, когда в комплексе имеется только одна хелатирующая группа SМе, а если две, то цирконий присоединяет два атома серы и делает все крайне энергично (рис.5, схема II). На это указывает одно обстоятельство: две освободившиеся метильные группы образуют реактив Гриньяра как побочный продукт реакции. Можно себе представить, сколь энергетически выгодно присоединение цирконием двух атомов серы, если при этом еще образуется исключительно реакционноспособное вещество.
Если хелатирующей группой служит NМе2, цирконий не отрывает метильную группу от азота, а разрывает связь С–Н в одной из них (рис.5, схема III). Это уже довольно близко к поставленной цели — разрыву С–Н-связи в углеводородах, но разница все же есть.
Наиболее интересные превращения мы наблюдали, когда стали восстанавливать комплексы, в которых хелатирующими группами были РR2 (R — Me или Ph). Цирконий не привлекал для своего окисления метильные остатки, связанные с фосфором, а использовал находящуюся рядом Ме-группу из циклопентадиенильного лиганда, разорвав в ней связь С–Н, в результате чего неожиданно возникла связь С–Zr (схема IV, соединение 1). Это уже разрыв связи в “настоящей” метильной группе, такая входит в состав углеводорода. Здесь хелатная группа работает по прямому назначению — заполняет координационную сферу металла.
Когда у фосфора расположены фенильные остатки, помимо соединения 1 (схема IV) образуется еще соединение 2 из-за отрыва атома водорода от фенильного радикала, что также интересно и неожиданно.
Комплексы Zr(II), показанные на схемах I—IV, неустойчивы и переходят в соединения Zr(IV). Тем не менее, комплекс с Zr(II) нам все же удалось получить, использовав для восстановления соединение с двумя группами РМе2 (рис.5, схема V). Теперь, когда поведение восстановленного циркония нам в общих чертах стало понятно, мы смогли объяснить этот факт. Как видно из рис.5, метильные остатки, с которыми цирконий не реагирует, те, что связаны с фосфором, а привлечь для взаимодействия Ме-группу из циклопентадиенильного лиганда мешают блокирующие его четыре метильные радикала, принадлежащие двум атомам фосфора (PMe2).
Итак, цирконий вполне успешно начинает вторгаться в область, где уверенно лидировали благородные металлы, и при этом демонстрирует большие возможности: восстановленные комплексы циркония могут разрывать связи O–C, S–C, Cалкил–H и Cарил–H. В этом весьма представительном наборе не хватает, пожалуй, только связи С–С, но мы полагаем, что сумеем разорвать и ее. Гораздо важнее другое — научиться проводить такие превращения не с группами, входящими в состав комплекса, т.е. не внутримолекулярно, а с реагентами, которые приближаются к комплексу извне.
Таким образом, подтверждается то, о чем мы говорили в самом начале: выполненная научная работа не бывает полностью завершенной, полученные результаты как бы сами формулируют новые задачи. А это вносит некоторый азарт в исследование и делает его интереснее.
Работа выполнена при поддержке Российского фонда фундаментальных исследований.
Проект 98-03-33057.Литература
1. Comprehensive coordination Chemistry / Ed. G.Wilkinson. Oxford; N.Y.; Frankfurt; San Paulo; Tokyo; Toronto. 1987. V.6.
2. Перевалова Э.Г., Решетова М.Д., Грандберг К.И.. Методы элементоорганической химии // Ферроцен. М, 1983.
3. Krut’ko D.P., Borzov M.V., Kuz’mina L.G. et al. // Inorg. Chim. Acta. 1998. V.280. №1—2. P.257—263.
4. Krut’ko D.P, Borzov M.V., Veksler E.N. et al. // Polyhedron. 1998. V.17. №22. P.3889—3901.
5. Krut’ko D.P., Borzov M.V., Veksler E.N. et al. // Eur. J. Inorg. Chem. 1999. №11. P.1973—1979.
6. Krut’ko D.P, Borzov M.V., Veksler E.N. et al. // Pure Appl. Chem. 2001. V.73. №2. P.367—371.